基础教程¶
主要讲述PyMOL常见的命令操作。
注解
1.本节教程是有顺序的。 2.建议先阅读入门教程,再阅读基础教程。 3.所有的命令都支持tab键补全,提高效率。 4. 主要的示例以Bace-1蛋白6EQM为例,里面含有配体小分子BUH。
命令输入窗口¶
PyMOL中有2个地方可供命令输入,如下图所示,根据自己的爱好,选择一处输入命令就可以了。
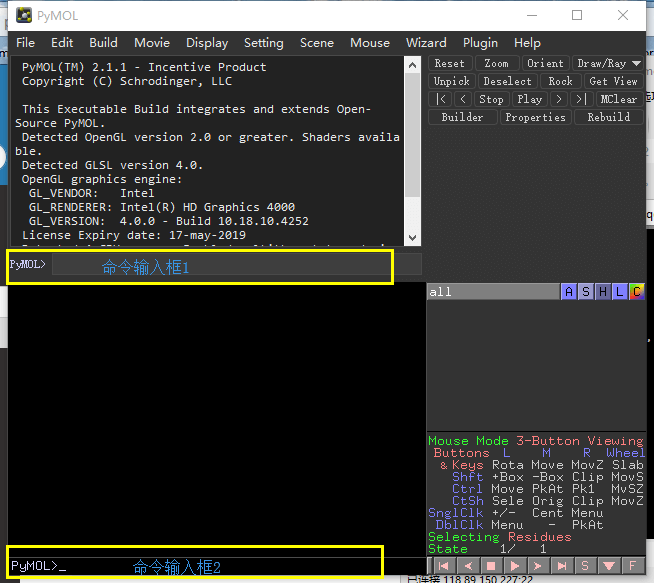
注解
注意可以把PyMOL当成一个python的模块(conda 安装 pymol 模块就可以),这样也可以在python脚本中使用。
注解
所见即所得是pymol的一个特色,所有鼠标操作都可以录制成命令。 参见PyMOL高级教程 宏录制 。
注解
输入命令,请用英文输入法;
忘记命令的名称¶
注解
google 关键词“pymolwiki sort”
忘记命令的用法¶
注解
方法一: 输入 “help cmdname” 比如 “help load”, “help png” 方法二:google 关键词“pymolwiki sort”
笔者倾向于使用命令框1,下面我将讲解一些常用的命令:
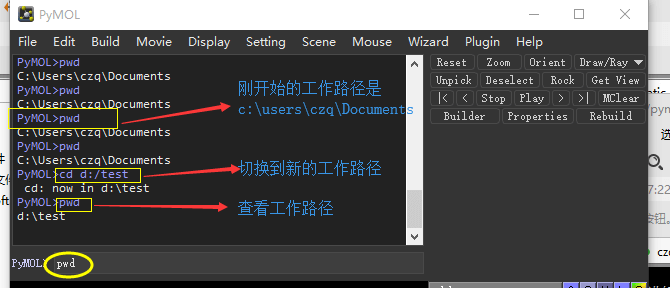
查看切换工作路径(pwd)¶
默认打开和保存文件,都是在工作路径中。因此设定合适的工作路径,可以提供效率。 建议分子文件格式,如mol2,pdb等格式,全部关联到PyMOL软件,不要关联到DS,Maestro等软件。
双击PDB文件,打开PyMOL软件,会自动调整工作路径为PDB文件所在路径。
- 命令 pwd; 查看工作路径;
- 命令 cd D:/test; 切换工作路径到D盘下面的test文件夹。在执行命令前,确保D盘下面有test 文件夹。
如图所示:
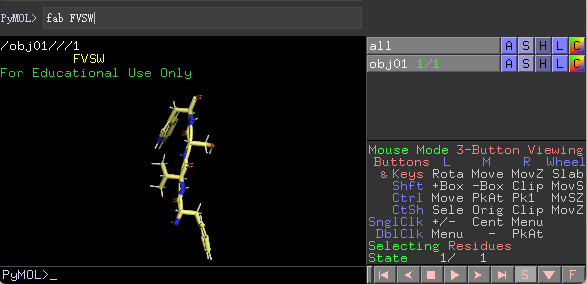
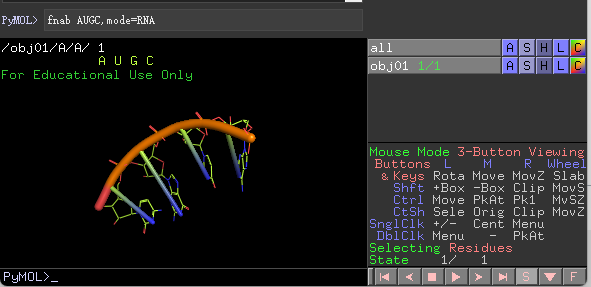
基于序列创建DNA/RNA (fnab)¶
比如需要创建一个双链ATGC的DNA,
直接在命令行窗口输入下面的命令就可以:
fnab ATGC
比如需要创建一个单链AUGC的RNA,
直接在命令行窗口输入下面的命令就可以:
fnab ATGC, mode = RNA
下载蛋白(fetch)¶
fetch 命令可以根据PDB ID编号下载蛋白结构到工作路径,并载入到PyMOL中显示,在PyMOL中的object的名字默认是PDB ID,可通过name参数进行修改。 从PyMOL 1.8开始,默认的格式是cif,可通过type参数修改文件的格式,也支持根据配体ligand ID的编号下载小分子。
示例:
- fetch 6FYZ # 下载编号是6FYZ的蛋白,文件格式是cif。
- fetch EBE # 下载编号是EBE的配体小分子,文件格式是cif。
- fetch 6MVD,name=”test”,type=pdb # 下载文件格式为pdb的6MVD的蛋白,载入PyMOL中设置名字为test.
type后面的参数不是字符串,不能加引号
type 支持的文件格式有:
默认是从pdb网站上下载数据的,可通过设置fetch_host 修改下载源。
set fetch_host,pdb set fetch_host,pdbe set fetch_host,pdbj
pdb 数据库有2个镜像库,一个是欧洲的pbde,另一个是日本的pdbj。 如果其中一个源不能使用的时候,可以尝试切换到其他源。
载入蛋白(load)¶
从PDB网站上下载蛋白,如 4hbk.pdb。 工作路径中已经存在PDB文件,如4hbk.pdb。 可通过load 命令进行加载。 命令:
load 4hbk.pdb load filename.pdb
load 和 fetch 的区别,load 是指定文件的名字,而fetch是分子的ID号。
载入amber轨迹文件 load¶
这里我以rst7文件为例
pymol 支持amber的trj(rst7) 格式和top格式 。
load in.prmtop,mol
load in.rst7,mol
设置surface的颜色(cartoon_color)¶
设置surface的颜色为red
set surface_color,red
注解
注意颜色是有优先级的,默认设置的是atom的颜色,如果没有设置surface的颜色,则继承atom的颜色
体验下
set surface_color,red
color blue
unset surface_color
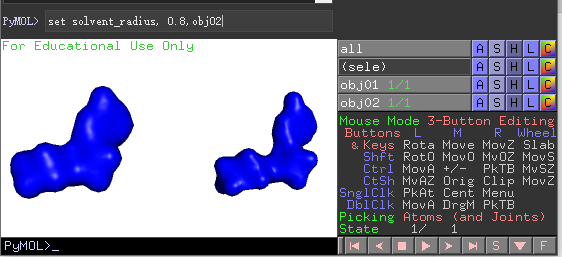
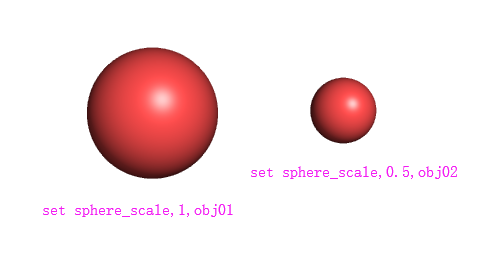
设置surface的大小(solvent_radius)¶
设置surface的大小,把surface设置细一点
set solvent_radius, 1.6
set solvent_radius, 0.8,obj02
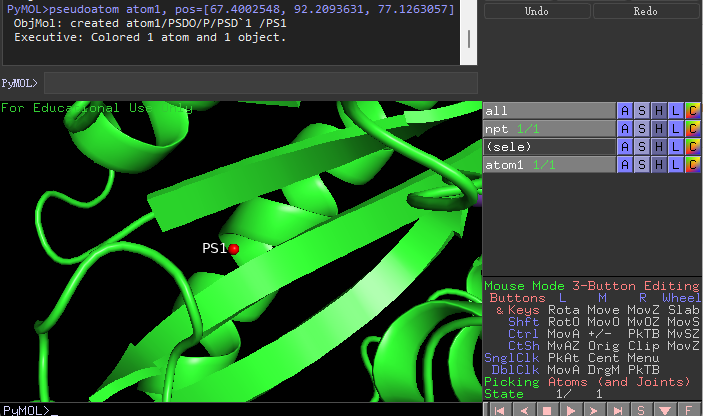
创建虚拟原子pseudoatom¶
比如计算口袋的质心坐标是 67.4002548, 92.2093631, 77.1263057,对其进行可视化检查。
pseudoatom atom1, pos=[67.4002548, 92.2093631, 77.1263057]
更多pseudoatom
设置截面的颜色¶
set ray_interior_color, grey
set ray_interior_color, grey,obj01
开关单双键模式 valence¶
示例 打开PyMOL fetch BUH bg_clor white set valence,1 set valence,0
效果如下:
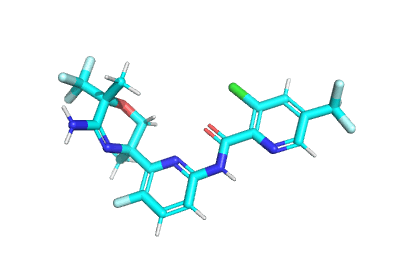
Fig 1. 双键模式 |
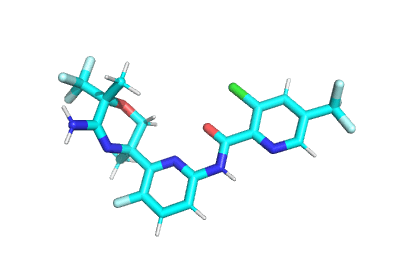
Fig 2. 普通模式 |
设置相互作用的样式¶
- 怎么像这样把相互作用的线调粗?
set dash_radius, 0.3
- 把虚线变成实线?
set dash_gap,0
- 设置实线区域的长度
set dash_length, 0.8
保存蛋白序列¶
save xxx.fasta,xxx
复制pymol 中特定的序列
fetch 4bhk
save 1.fasta, 4bhk and chain A and resi 200-210
高清图片(ray)¶
制作高清图片, 制作完图片后输入ray命令,然后保存图片。
设置两个光源 two_sided_lighting¶
set two_sided_lighting,1
当蛋白表面有破损的时候,ray的时候可能会出现黑洞现象,如下图所示。
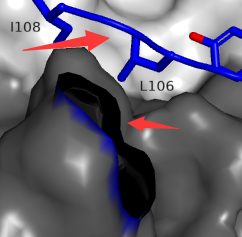
使用上述命令,设置两个光源就可以消除黑洞现象。
添加氨基酸残基编号¶
label obj01 and name ca, "%s-%s"%(resn,resi)
设置label的小数位数¶
默认距离和角度是1位,通过下面命令可以修改,显示的位数。
设置label的字体大小¶
label字体的默认大小是14pt, 单位是pt。
除了使用pt单位,也可以使用Angstrom 单位,如设置字体大小为 1.2 Angstrom。
置顶label¶
注解
float_labels (boolean, default: off) controls whether or not labels are drawn last without Z-buffer clipping so that they appear to float over the image.
This setting has no effect when ray tracing.
利用 float_labels 命令可以将标签置顶,浮动到分子表面。
默认是 set float_labels,0
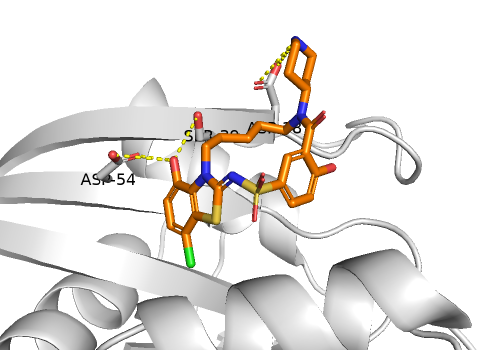
修改成 set float_labels,1,这样就可以看到感兴趣的残基的编号,效果如下:
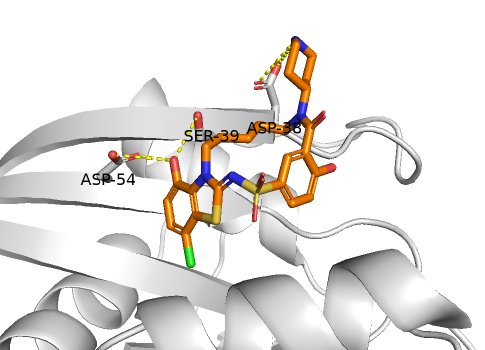
我们可以看到感兴趣的氨基酸残基是ASP38/SER39/ASP54 。
注解
ray的时候这个效果会消失,可尝试使用**draw**命令进行渲染。 如果还达不到想要的要求,可以在ppt中手动添加label。
保存object¶
cmd.save(filename[, selection[, state[, format]]])
save file [,(selection) [,state [,format]] ]
PyMOL>save 01.pdb, ONLYprotein,
Save: wrote “01.pdb”.
PyMOL>cmd.save(“0.pdb”,”ONLYprotein”)
掩藏object-disable¶
disable objectName
掩藏representation¶
hide stick hide cartoon
切换编辑模式¶
edit_mode
蛋白的展现形式(show,as)¶
命令如下:
默认的object name是all。
示例讲解: 打开pymol, 依次执行下面命令。 1. cd d:PyMOLstartedManualpse 切换到工作路径 2. fetch 6EQM 下载蛋白 3.
设置双键的显示形式 valence¶
set valence, 0 #关闭双键显示
set valence, 1 #开启双键显示
设置stick的粗细¶
set stick_radius, 0.12
重新计算二级结构(dss)¶
dss
选择中的关键词和缩略¶
- organic org. Non-polymer organic compounds (e.g. ligands, buffers)
- inorganic ino. Non-polymer inorganic atoms/ions
- solvent sol. Water molecules
- polymer pol. Protein or Nucleic Acid
- polymer.protein Protein (New in PyMOL 2.1)
- polymer.nucleic Nucleic Acid (New in PyMOL 2.1)
- guide Protein CA and nucleic acid C4*/C4’
- hetatm Atoms loaded from PDB HETATM records
- hydrogens h. Hydrogen atoms
- backbone bb. Polymer backbone atoms (new in PyMOL 1.6.1)
- sidechain sc. Polymer non-backbone atoms (new in PyMOL 1.6.1)
- metals Metal atoms (new in PyMOL 1.6.1)
- donors don. Hydrogen bond donor atoms
- acceptors acc. Hydrogen bond acceptor atoms
cartoon 模式¶
pymol 中内置了10种cartoon 模式:
- skip (-1) 不显示
- automatic (0) (use ss property) 默认模式
- loop (1)
- rectangle (2)
- oval (3)
- tube (4)
- arrow (5)
- dumbbell (6) (see also cartoon_fancy_helices)
- putty (7) (b-factor scaling)
- dash (8) (new in PyMOL 1.8.2)
在命令行窗口输入下面的命令对各种模式进行感受。 .. code-block:: console
cartoon loop cartoon tube cartoon putty
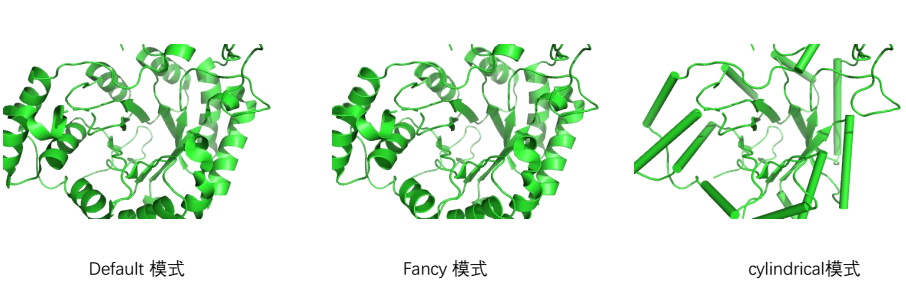
cartoon helix 模式¶
在cartoon automaic 模式下 pymol 中内置了3种cartoon helix模式:
- 默认default模式
- Fancy Helices 模式 两端是尖尖
- Cylindrical Helices 模式 圆柱体
开启 Fancy 模式
set cartoon_fancy_helices, 1
开启 cylindrcal 模式
set cartoon_cylindrical_helices, 1
设置圆柱的粗细:
set cartoon_helix_radius,0.9
set cartoon_helix_radius,2.5
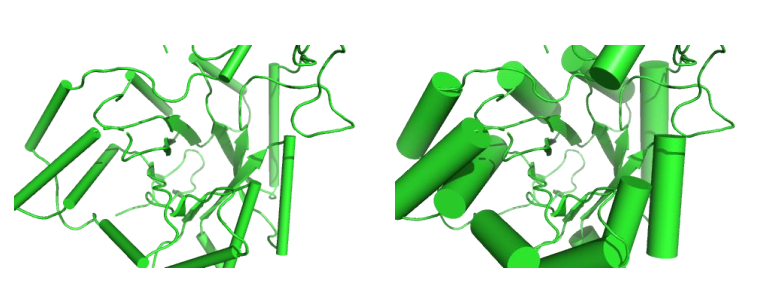
启用默认 default 模式
set cartoon_fancy_helices, 0
set cartoon_cylindrical_helices, 0
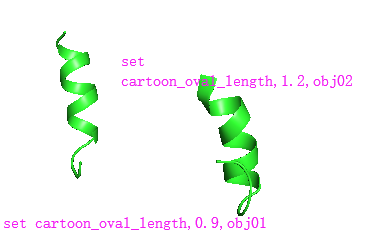
定义fancy 和default模式下的α-helix¶
命令:
set cartoon_oval_length , 0.8 # default is 1.20
set cartoon_oval_width , 0.2 # default is 0.25
示例:
着色(color)¶
color red,object name
设置透明度(transparency)¶
所有的命令
选择,残基(select)¶
选择所有的碳原子: select catoms, elem C
选择蛋白:select protein, (byres polymer & name CA)
选择核酸: select nucleic, (byres polymer & name P)
选择RNA: select rna, (byres polymer & name O2’)
选择DNA: select dna, (nucleic & !rna)
选择小分子周围3A的氨基酸残基: select pocket3A, byres (resn ligName around 3 and objectName)
选择小分子周围4A的氨基酸残基: select pocket3A, byres (resn ligName around 4 and objectName)
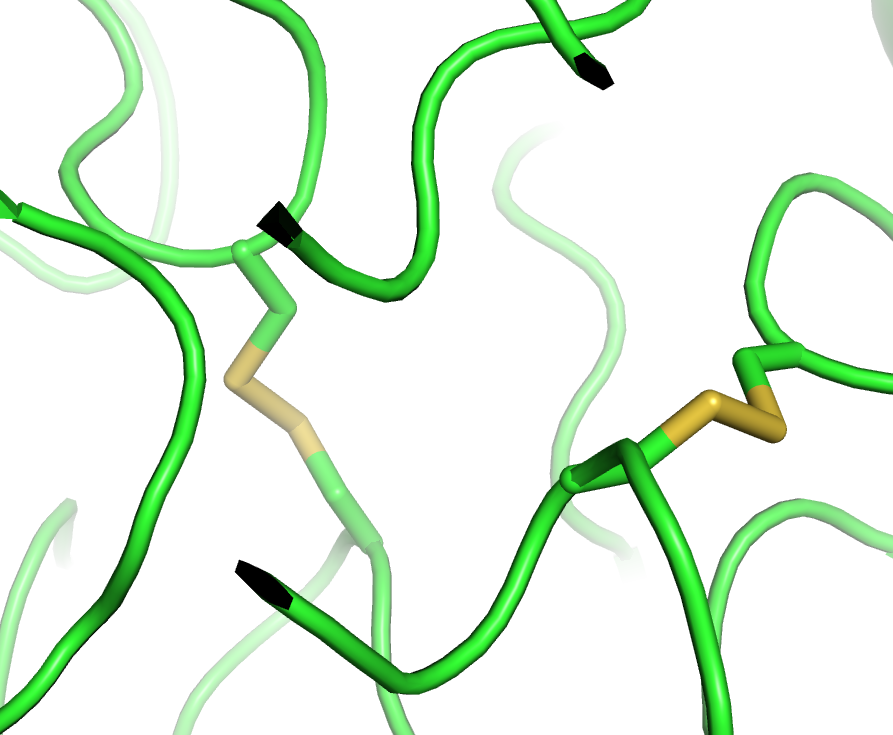
查看蛋白中的二硫键¶
显示二硫键为黄色stick模式
show sticks, (cys/ca+cb+sg) and byres (cys/sg and bound_to cys/sg)
创建二硫键
create disulfide_bonds, (cys/ca+cb+sg) and byres (cys/sg and bound_to cys/sg)
注解
如果PDB文件是amber格式,还原态是CYS,二硫键是CYX,去质子化是CYM。 show sticks, (cyx/ca+cb+sg) and byres (cyx/sg and bound_to cyx/sg)
计算蛋白或者小分子的分子量¶
鼠标版本:
A->compute->molecule weight
命令行(脚本)版本:
from pymol import util
cmd.load("4hbk.pdb")
util.compute_mass("4hbk",implicit=True,quiet=0,_self=cmd)
计算表面积get_area¶
利用 get_area 计算表面积,支持分子表面积和 solvent accessible surface area(SASA)表面积。
比如计算整个蛋白的分子表面积
注解
get_area all
比如计算整个蛋白的SASA表面积
注解
set dot_solvent, 1
get_area all
计算66号氨基酸的表面积
注解
get_area resi 66
计算66号氨基酸的SASA表面积
注解
set dot_solvent, 1
get_area resi 66
显示蛋白的口袋的静电势表面¶
step1. 显示整个蛋白蛋白的静电势表面;A->generate->vacuum electrostatic->potential
step2 设置口袋的中心,一般以其中的配体作为中心;
set surface_carve_selection, ligand
step3 设置口袋的大小
set surface_carve_cutoff, 5
step4 设置口袋的表面的透明度
set transparency,0.3, 1azm_e_chg
step5 切换为溶剂可接触表面
set surface_solvent, 1
删除配体分子5A以外的水分子¶
remove resn hoh and (resi 307 gap 5)
删除极性氢原子¶
remove obj01 & hydro & not nbr. (don.|acc.)
cmd.remove("obj01 & hydro & not nbr. (don.|acc.)")
叠合两个object(super)¶
参考: https://pymolwiki.org/index.php/Super
自动进行全局叠合
super 会把两个object进行叠合,和align的区别主要是
- super 主要是基于机构进行叠合;
- align 主要是基于序列进行叠合。
当两个蛋白的序列相似度较低的时候,使用super叠合的效果比align叠合的效果好。
super p1, p2
指定叠合部分
super p1 & alt A+'', p2 & alt B+''
实战
super obj03 & resi 2-191 & chain K, obj04 & resi 2-191 & chain C
分组(group)¶
group efHand, 1cll 1ggz 1sra
group efHand, open
group efHand, close
创建嵌套的组
group target,pdbid
group surf, *_e_*
group target,surf
排序object (order)¶
order object
颜色的名字¶
PyMOL 中内置超过`100种颜色 <https://pymolwiki.org/index.php/Color_Values>`_,
aquamarine 0.5 1.0 1.0
black 0.0 0.0 0.0
blue 0.0 0.0 1.0
bluewhite 0.85 0.85 1.00
br0 0.1 0.1 1.0
br1 0.2 0.1 0.9
br2 0.3 0.1 0.8
br3 0.4 0.1 0.7
br4 0.5 0.1 0.6
br5 0.6 0.1 0.5
br6 0.7 0.1 0.4
br7 0.8 0.1 0.3
br8 0.9 0.1 0.2
br9 1.0 0.1 0.1
brightorange 1.0 0.7 0.2
brown 0.65 0.32 0.17
carbon 0.2 1.0 0.2
chartreuse 0.5 1.0 0.0
chocolate 0.555 0.222 0.111
cyan 0.0 1.0 1.0
darksalmon 0.73 0.55 0.52
dash 1.0 1.0 0.0
deepblue 0.25 0.25 0.65 deep
deepolive 0.6 0.6 0.1
deeppurple 0.6 0.1 0.6
deepsalmon 1.0 0.5 0.5 duplicated?
deepsalmon 1.00 0.42 0.42
... ...
violetpurple 0.55 0.25 0.60
warmpink 0.85 0.20 0.50
wheat 0.99 0.82 0.65
white 1.0 1.0 1.0
yellow 1.0 1.0 0.0
yelloworange 1.0 0.87 0.37
设置二级结构的颜色¶
PyMOL中内置了3种二级结构着色策略。
helix sheet loop
red yellow green (策略1)
cyan magenta salmon (策略2)
cyan red magenta (策略3)
除了上述内置的着色策略,我们还可以自定义二级结构的颜色。
方法一:
选定二级结构对应的氨基酸残基,然后设置颜色。
方法二(推荐):
color red, ss h
color yellow, ss s
color green, ss l+''
推荐一套配色策略,感觉比内置的好看一点。
PyMOL>color yellow,ss s
PyMOL>color grey,ss l+''
PyMOL>color teal,ss h
安装rdkit¶
PyMOL>assign_stereo %obj01, 1
Method "rdkit" not available (No module named 'rdkit')
Method "schrodinger" not available (Error: SCHRODINGER environment variable not set)
Error: need SCHRODINGER or rdkit
Error: please install rdkit or set SCHRODINGER variable
定位到PyMOL的安装目录,用安装目录中的conda 安装rdkit; 注意: 在windows下用cmd窗口,不要使用 powershell 窗口。 先激活base环境,然后安装rdkit。
Microsoft Windows [版本 10.0.22621.2134]
(c) Microsoft Corporation。保留所有权利。
C:\Users\Lenovo>d:
D:\>cd PYMOL3/
D:\PYMOL3>cd condabin
D:\PYMOL3\condabin>conda.bat install rdkit -c conda-forge
Channels:
- conda-forge
- defaults
Platform: win-64
Collecting package metadata (repodata.json): done
Solving environment: done
## Package Plan ##
environment location: C:\Users\Lenovo\AppData\Roaming\SVINA\miniconda
added / updated specs:
- rdkit
The following packages will be downloaded:
package | build
---------------------------|-----------------
_openmp_mutex-4.5 | 2_gnu 48 KB conda-forge
brotli-1.1.0 | h2466b09_3 20 KB conda-forge
brotli-bin-1.1.0 | h2466b09_3 21 KB conda-forge
