高级教程¶
主要讲述PyMOL中脚本的编写、命令的自定义、插件的编写、插件的介绍。
PyMOL脚本、命令、插件¶
PyMOL支持Python 编程语言,故可以借助python让pymol无所不能,pymol除了显示软件,也能成为计算软件。
脚本:把pymol作为一个模块,实现一些计算功能。 命令:pymol中内置了一些命令,如color、dist等,我们也可以自定义新的命令。 插件:pymol中内置了一些插件,如apbs-pdb2pqr等,我们也可以自定义插件。
注解
编写PyMOL脚本、命令、插件,需要有一定的python基础。python教程:HOW2PY
安装PyMOLwiki中的命令¶
这里我以这个命令 `FocalBlur<https://pymolwiki.org/index.php/FocalBlur>`_ 为例进行演示。
- 下载 focal_blur.py脚本,https://raw.githubusercontent.com/Pymol-Scripts/Pymol-script-repo/master/focal_blur.py 比如下载到D盘根目录下面 d:/focal_blur.py
- 打开pymol,在命令窗口输入run d:/focal_blur.py 就完成了安装。
- 运行命令 FocalBlur aperture=4,samples=400,ray=0
FocalBlur一共有5个参数:
- aperture
- samples
- ray
- width
- height
该命名和单反的光圈相关,控制景深的。 需要开启ray=1 才能看到光圈景深的效果
修改PyMOL的默认设置¶
我个人习惯保存突变的时候使用白色背景、关闭光照阴影,而默认的是透明背景, 每次保存图片的时候都需要用命令” set ray_opaque_background, 1 “设置白色背景,比较麻烦。 我们可以通过下面的方法,修改PyMOL的默认设置。
建议创建一个脚本 pymolset.py, 假设该脚本存放在D盘
# AUTOGENERATED FILE
from pymol import cmd, invocation
cmd.set("bg_rgb", 'white')
cmd.set("ray_opaque_background", '1')
cmd.set("ray_shadow", 'off')
如果不知道如何定义pymolset.py,可以通过 save_settings.py
如果无法下载从国外网站下载,可通过下面的百度云链接进行下载。
链接:https://pan.baidu.com/s/1vWNU5ssyOTvMkPAHVqQ27g?pwd=ejsg
提取码:ejsg
在PyMol 命令窗口下,先修改成自己想要的设置,运行下面的命令就可以自动生成pymolset.py 文件。
run save_settings.py
save_settings pymolset.py
生成pymolset.py后,用文本编辑器打开进行编辑修改,保留自己需要设置就可以了。
创建命令¶
这里我举一个例子: 计算两个苯环之间的距离或者五元环和苯环之间的距离。
拓展:计算2个object的中心距离。
首先我们为命令起名字,要求是简洁高效,通过命令就能猜测到这个命令的功能。 这里我起的脚本名 dist_center。 核心是在python 脚本中定义这个函数,并cmd.extend拓展命令。 框架如下
1 2 3 4 | from pymol import cmd
def dist_center(obj1="obj01",obj2="obj02",showflag=True):
pass
cmd.extend("dist_center", dist_center)
|
完整代码(dist_center_2obj.py)如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 | """
DESCRIPTION
measure the distance between the centers of two objects
the object usually is benzene, pyridine and other cyclo object
more information at:
http://pymol.chenzhaoqiang.com/intro/advanceManual.html
AUTHOR
Zhaoqiang Chen, 2017
Please inform me if you use/improve/like/dislike/publish with this script.
email: 744891290@qq.com
"""
from pymol import cmd,math
def getcenterofatoms(atoms):
'''
'''
x=0
y=0
z=0
for atm in atoms:
x+=atm.coord[0]
y+=atm.coord[1]
z+=atm.coord[2]
numatm=float(len(atoms))
x=x/numatm
y=y/numatm
z=z/numatm
return [x,y,z]
def getdist(cen1,cen2):
'''
'''
dx=cen1[0]-cen2[0]
dy=cen1[1]-cen2[1]
dz=cen1[2]-cen2[2]
dist=dx*dx+dy*dy+dz*dz
return math.sqrt(dist)
def dist_center(obj1="obj01",obj2="obj02",showflag=True):
'''
usage1:
dist_center obj01,obj02
usage2:
dist_center obj01,obj02,1
usage3:
dist_center obj01,obj02,0
usage1 is equal to usage2!
'''
obj1atoms=cmd.get_model(obj1).atom
obj2atoms=cmd.get_model(obj2).atom
obj1center=getcenterofatoms(obj1atoms)
obj2center=getcenterofatoms(obj2atoms)
dist = getdist(obj1center,obj2center)
print("distance: %.2f"%(dist ))
if showflag:
cmd.pseudoatom('cen1', pos=obj1center)
cmd.pseudoatom('cen2', pos=obj2center)
cmd.distance("dist","cen1","cen2")
cmd.extend("dist_center", dist_center)
|
演示如下:
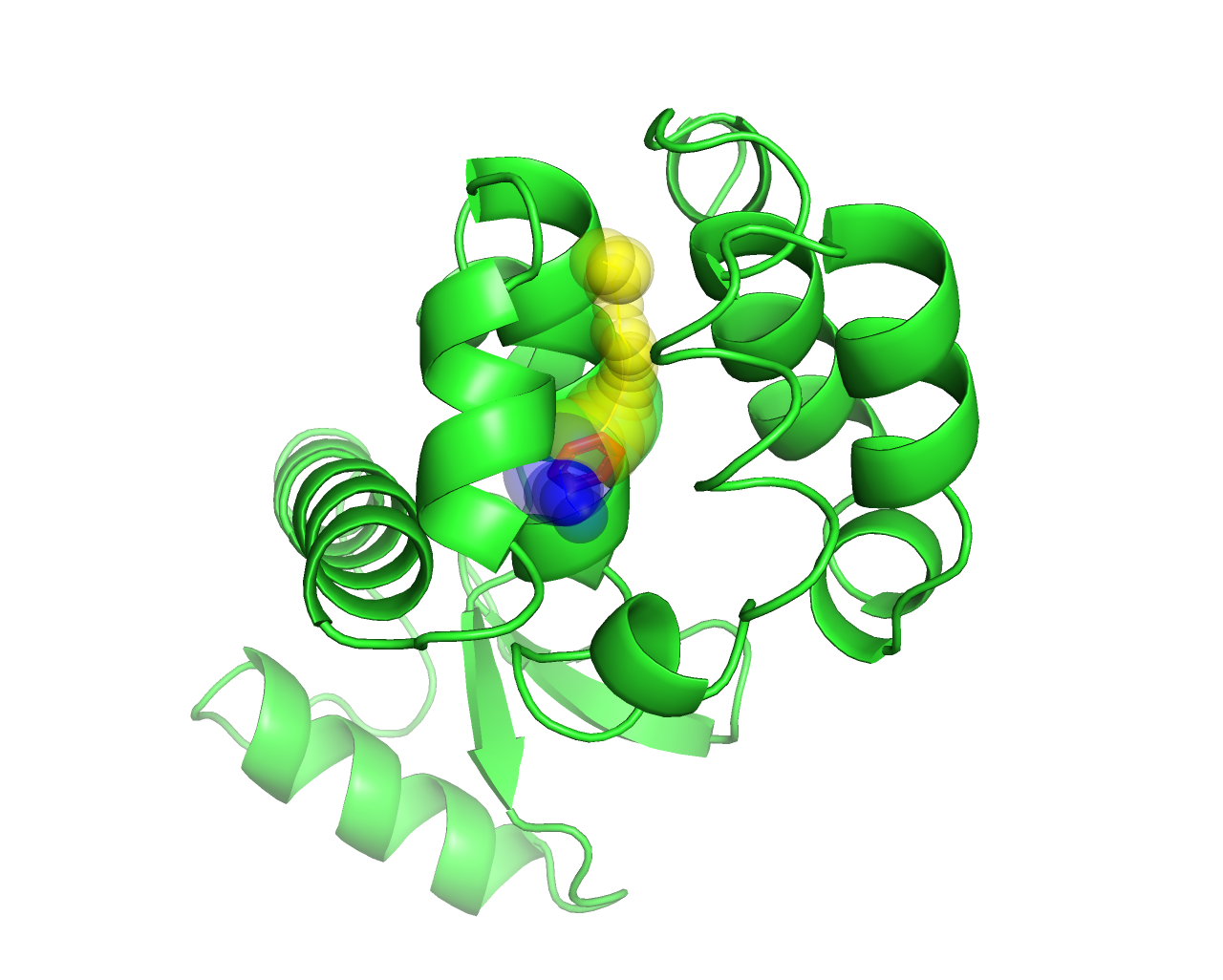
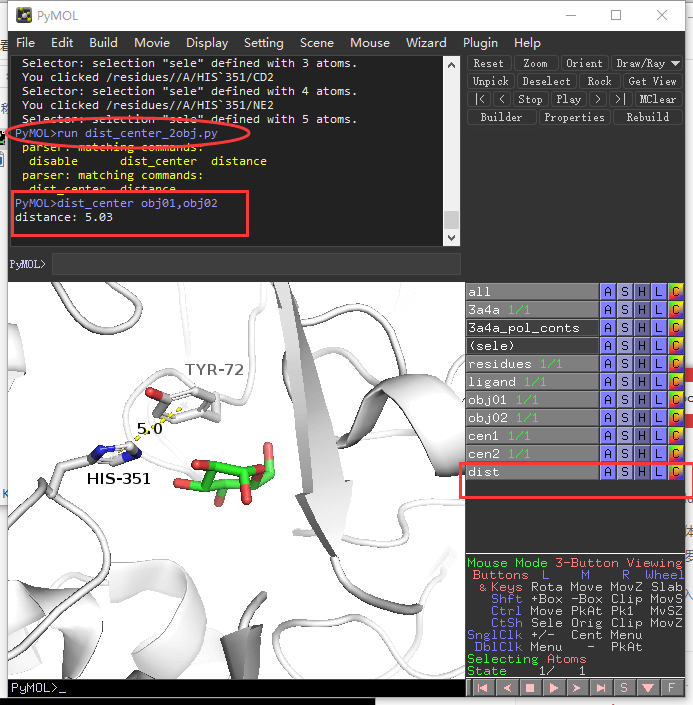
载入蛋白,运行run dist_center_2obj.py命令; 把TRY-72侧链苯环6个原子保存为obj01; 把HIS-351侧链咪唑环的5个原子保存为obj02; 运行命令 dist_center obj01,obj02 即可测量距离。
计算并标注氨基酸残基的表面积¶
计算并标注object上的每个氨基酸残基的表面积。
下载脚本(右击另存为):labelSASA.py 。
from pymol import cmd
def labelSASA(selection='(all)'):
'''
DESCRIPTION
Radius of gyration
calculate the MS or SASA of per residues on the selection object
USAGE
labelSASA [ selection ]
'''
# set dot_solvent, on
# mode = int(mode)
# cmd.set("dot_solvent",mode)
atoms = cmd.get_model(selection).atom
total_sa=0
for atom in atoms:
if atom.name =='CA':
res_per="chain %s and resi %s and resn %s"%(atom.chain,atom.resi,atom.resn)
sa = "%.2f"%cmd.get_area( res_per)
print(res_per,sa)
total_sa+=float(sa)
res_per_CA="chain %s and resi %s and resn %s and name CA"%(atom.chain,atom.resi,atom.resn)
cmd.label(res_per_CA,sa)
print("total surface area",total_sa)
return total_sa
cmd.extend("labelSASA", labelSASA)
运行
下载脚本后,文件名修改为labeSASA.py
run labeSASA.py
# 在pymol中选择感兴趣的残基,比如所有的cys, select cyss,resn cys
select cyss,resn cys
labelSASA cyss
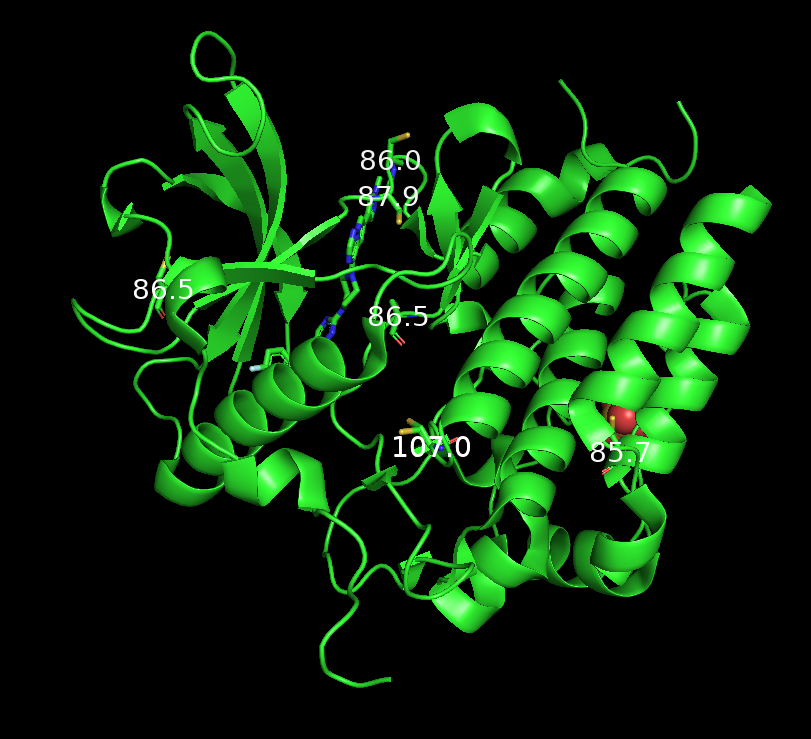
输出:
PyMOL>run labelSASA.py
PyMOL>select cyss,resn cys
Selector: selection "cyss" defined with 39 atoms.
PyMOL>labelSASA cyss
chain C and resi 660 and resn CYS 86.47
chain C and resi 673 and resn CYS 87.92
chain C and resi 674 and resn CYS 86.04
chain C and resi 788 and resn CYS 106.99
chain C and resi 788 and resn CYS 106.99
chain C and resi 809 and resn CYS 86.47
chain C and resi 906 and resn CYS 85.66
total surface area 646.54
注解
参数说明:
默认是普通表面积计算 SASA表面积计算,set dot_solvent, on
创建向量draw_vector¶
拉伸动力学需要创建向量,对向量进行可视化检查。
下载脚本(右击另存为):addproteinBox.py 。
运行
run draw_vector.py
draw_vector 67,92,77,66,92,76
注解
参数说明: 命令draw_vector,向量起点坐标x,向量起点坐标y,向量起点坐标z,向量终点坐标x,向量终点坐标y,向量终点坐标z
编写脚本: draw_vector.py
from pymol import cmd, cgo
from pymol.cgo import *
# 全局变量,用于跟踪向量数量
vector_counter = 1
def draw_vector(x1, y1, z1, x2, y2, z2):
global vector_counter # 引入全局计数器
# Convert the arguments to floats to ensure they are real numbers
x1, y1, z1, x2, y2, z2 = float(x1), float(y1), float(z1), float(x2), float(y2), float(z2)
# 定义箭头头部的大小
arrow_radius = 0.3 # 箭头底部的半径
arrow_length = 0.5 # 箭头的长度
cylinder_radius = 0.1 # 圆柱的半径
# 计算箭头起点 (箭头的尾部,也就是圆柱的末端)
vec_x = x2 - x1
vec_y = y2 - y1
vec_z = z2 - z1
norm = (vec_x**2 + vec_y**2 + vec_z**2)**0.5
# 箭头尾端位置 (让箭头稍微缩短一段)
arrow_base_x = x2 - (arrow_length * vec_x / norm)
arrow_base_y = y2 - (arrow_length * vec_y / norm)
arrow_base_z = z2 - (arrow_length * vec_z / norm)
print("coord1",x1,y1,z1)
print("coord2",x2,y2,z2)
print("middle",arrow_base_x, arrow_base_y, arrow_base_z)
# 定义CGO对象(圆柱 + 箭头)
vector_cgo = [
# 箭头部分 (CONE)
cgo.CONE, arrow_base_x, arrow_base_y, arrow_base_z, x2, y2, z2, arrow_radius, 0.0,
1.0, 0.0, 0.0, # 箭头底部颜色 (红色)
1.0, 0.0, 0.0,# 箭头顶端颜色 (红色)
1.0, 1.0, # 封闭圆锥底部
# 圆柱体部分
cgo.CYLINDER, x1, y1, z1, arrow_base_x, arrow_base_y, arrow_base_z, cylinder_radius,
1.0, 0.0, 0.0, # 起点颜色 (红色)
1.0, 0.0, 0.0, # 终点颜色 (红色)
# w = 0.06 # cylinder width
# l = 0.75 # cylinder length
# h = 0.25 # cone hight
# d = w * 1.618 # cone base diameter
# cgo.CONE, l, 0.0, 0.0, h+l, 0.0, 0.0, d, 0.0,
# 1.0, 0.0, 0.0,
# 1.0, 0.0, 0.0,
# 1.0, 1.0,
]
# 使用递增的名字
obj_name = "vector_%03d" % vector_counter
vector_counter += 1 # 递增计数器
# 加载CGO对象
cmd.load_cgo(vector_cgo, obj_name)
# 扩展 PyMOL 命令,使其支持 draw_vector
cmd.extend("draw_vector", draw_vector)
更多PyMol CGO定义:
注解
注意一定要严格按照PyMol CGO规则进行定义。 和常见的CGO语法不完全一致。
回转半径(Radius of gyration)¶
蛋白从apo构象变成holo构象,通常会伴随着Rog的变化。 如果结合的更紧密,则Rog会变小。 如果打开口袋,则Rog会变大。
from pymol import cmd
import math
def rgyrate(selection='(all)', quiet=1):
'''
DESCRIPTION
Radius of gyration
USAGE
rgyrate [ selection ]
'''
try:
from itertools import izip
except ImportError:
izip = zip
quiet = int(quiet)
model = cmd.get_model(selection).atom
x = [i.coord for i in model]
mass = [i.get_mass() for i in model]
xm = [(m*i,m*j,m*k) for (i,j,k),m in izip(x,mass)]
tmass = sum(mass)
rr = sum(mi*i+mj*j+mk*k for (i,j,k),(mi,mj,mk) in izip(x,xm))
mm = sum((sum(i)/tmass)**2 for i in izip(*xm))
rg = math.sqrt(rr/tmass - mm)
if not quiet:
print("Radius of gyration: %.2f" % (rg))
return rg
cmd.extend("rgyrate", rgyrate)
下载脚本(右击另存为):addproteinBox.py 。
运行
run rgyrate open.py
load open.pdb
load close.pdb
rgyrate open
rgyrate close
输出:
PyMOL>rgyrate close
Radius of gyration: 21.14
PyMOL>rgyrate open
Radius of gyration: 21.43
从apo 到holo 仅仅变化了1%, 不一定是一个好的CV。 10%以上可以尝试作为一个CV指标。
注解
参数说明: 命令rgyrate,object名字
基于states的号码导出分子¶
场景:
虚筛挑选分子的时候,记录感兴趣的state。
思路: 1. 需要保存的states转换成列表。 2. 迭代states进行保存。
代码文件 save_states.py :
from pymol import cmd
def save_states(obj,states=""):
'''
save_states obj01,states ="1,3,4"
'''
stats = []
states=states.replace('"','')
for i in states.split(','):
print(i)
i=int(i)
stats.append(i)
for i in stats:
newfilename="%s_%s.sdf"%(obj,i)
cmd.save(newfilename,obj,state=i)
cmd.extend("save_states",save_states)
使用说明:
run save_states.py
save_states obj01,states ="1,3,4"
显示蛋白残基之间的盐桥¶
思路:
- 先定位带负电荷的氨基酸上的羧基上的氧原子;
- 然后定位正电荷氨基的Lys 上的带正电荷的N原子;
- 最后显示这两类原子在4A范围之内的配对情况。
方法1 命令行版本
注解
PyMOL>select negative, (resn ASP+Glu and name OD*+OE*)
PyMOL>select positive, (resn Lys and name NZ) or (resn arg and name NE+NH*)
PyMOL>distance saltbridge, (negative ), (positive ), 4.0, 0
PyMOL>select negative, (resn ASP+Glu and name OD*+OE*)
PyMOL>select positive, (resn Lys and name NZ) or (resn arg and name NE+NH*)
PyMOL>distance saltbridge, (negative ), (positive ), 4.0, 0
方法2 拓展成一个命令 saltbridge.py
from pymol import cmd
def show_saltbridge(dist=4.0):
cmd.select("negative","resn ASP+Glu and name OD*+OE*")
cmd.select("positive","(resn Lys and name NZ) or (resn arg and name NE+NH*)")
cmd.distance("saltbridge","negative","positive",dist,0)
cmd.extend("show_saltbridge", show_saltbridge)
使用命令
PyMOL>run saltbridge.py
PyMOL>show_saltbridge 3
PyMOL>show_saltbridge
用法如图所示:
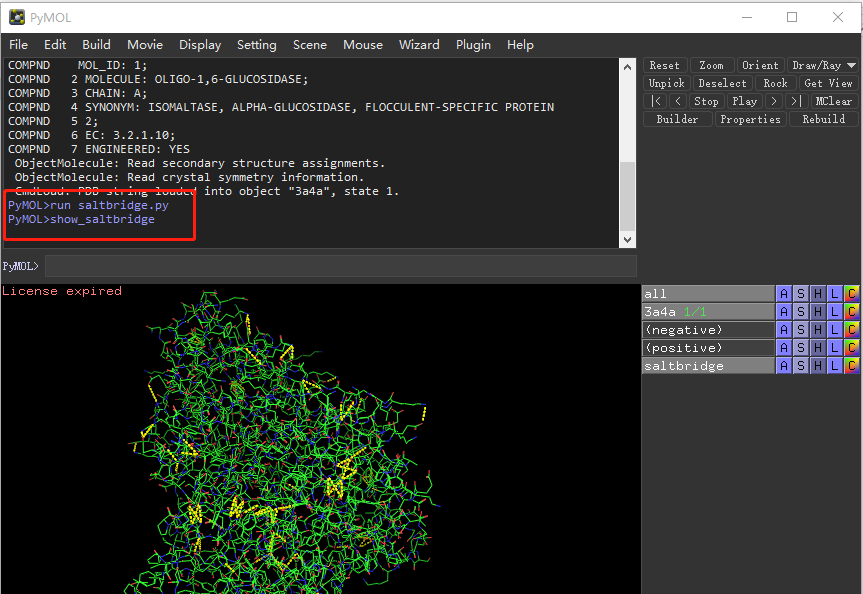
两个氨基酸之间存在黄色虚线,说明这个氨基酸存在盐桥。
基于选中的残基定义对接盒子¶
基于选中的残基计算对接盒子的中心
下载脚本(右击另存为) addproteinBox.py 。
在PyMOL 命令行,运行下述命令,就可以该蛋白添加盒子,并显示长宽高。
第一步:PyMOL>run center_box.py
第二步:选择感兴趣的氨基酸残基
第三步:PyMOL>center_box
结果:
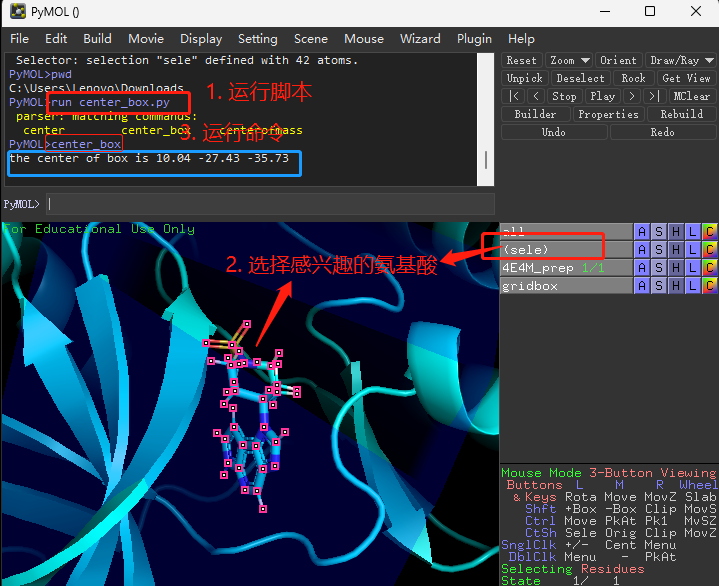
把蛋白装入盒子中,并可视化长宽高¶
计算蛋白的长宽高,并可视化。
下载脚本(右击另存为) addproteinBox.py 。
在PyMOL 命令行,运行下述命令,就可以该蛋白添加盒子,并显示长宽高。
PyMOL>run addproteinBox.py
PyMOL>add_proteinbox 4hbk
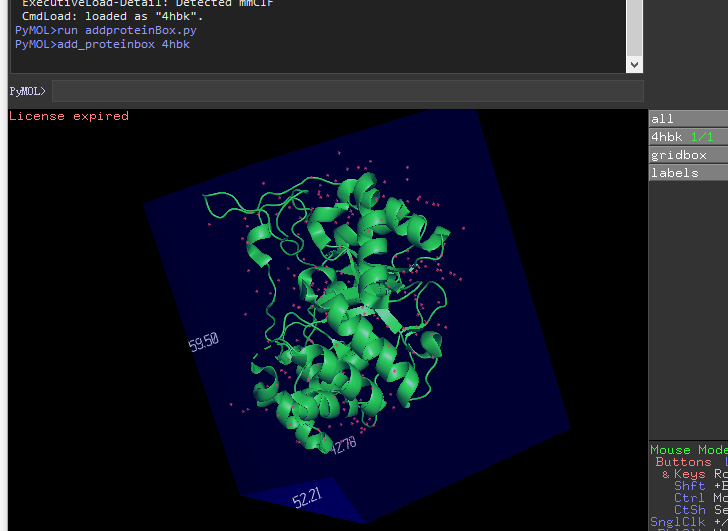
左下角显示XYZ坐标¶
VMD左下角默认会显示XYZ坐标,PyMOL也可以做到。
下载脚本(右击另存为) axes.py 。
在PyMOL 命令行,运行下述命令,就可以添加XYZ坐标了。
PyMOL>run axes.py
PyMOL>axes
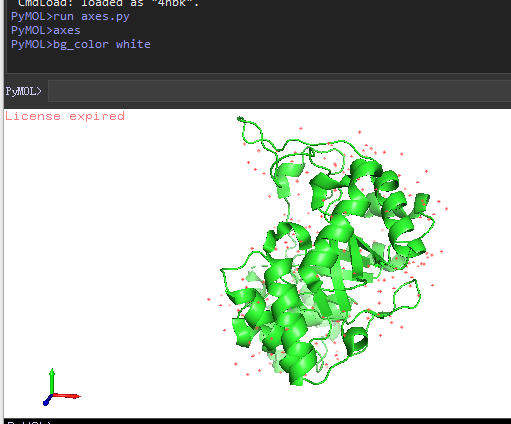
加载坐标颜色文件¶
文件(test.xyzrgb)格式如下
x y z r g b
下载笔者编写的脚本 loadxyzrgb_sub.py
在 PyMOL 命令行,运行下述命令,就可以可视化坐标颜色文件了。
PyMOL>run loadxyzrgb_sub.py
PyMOL>loadxyzrgb test.xyzrgb
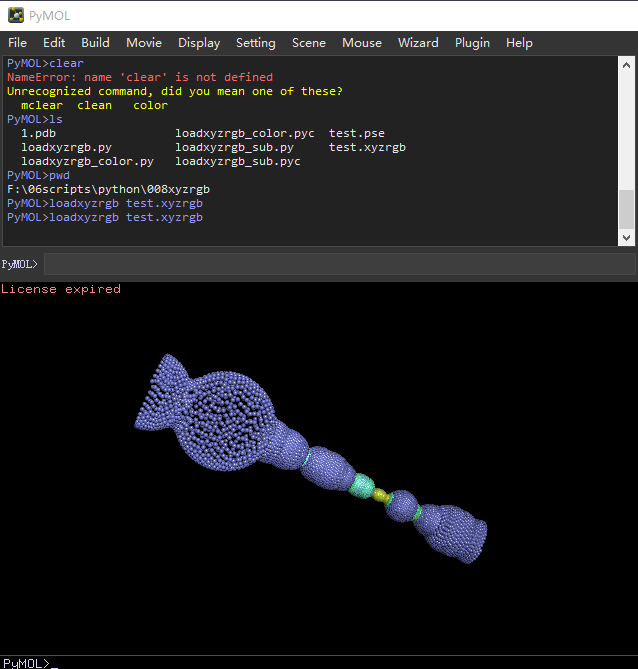
提取配体周围的位点¶
from pymol import cmd
from pymol import util
# example 6SP6 KJK A
def extractSiteByLig(pdbid,ligid,radius=4,chainid=None):
'''
'''
cmd.delete('all')
cmd.fetch(pdbid)
if not chainid:
cmd.select("ligall","resn %s"%ligid)
atms_lig = cmd.get_model("ligall").atom
chains_lig = [ atom.chain for atom in atms_lig ]
chains_lig = list(set(chains_lig))
chainid = chains_lig[0]
cmd.select("lig_%s"%chainid," chain %s and resn %s "%(chainid,ligid))
cmd.select("site","byres (lig_%s expand %s)"%(chainid,radius))
cmd.create("pocket","site")
cmd.disable("all")
cmd.enable("pocket")
cmd.do("as sticks,pocket")
util.cba(144,"pocket")
util.cba(120,"pocket and resn %s"%ligid)
newfile="%s_%s_%s_%s.pdb"%(pdbid,ligid,chainid,radius)
cmd.save(newfile,"site")
# extractSiteByLig 6SP6, KJK
cmd.extend("extractSiteByLig",extractSiteByLig)
pdbid = '6SP6'
ligid = 'KJK'
extractSiteByLig(pdbid,ligid)
突变mutate命令行插件¶
编写mutate命令插件脚本mutate.py
from pymol import cmd
#refer: https://pymolwiki.org/index.php/Rotkit
#qq group: 294266356
def mutate(molecule, chain, resi, target="CYS", mutframe="1"):
target = target.upper()
cmd.wizard("mutagenesis")
cmd.do("refresh_wizard")
cmd.get_wizard().set_mode("%s" % target)
selection = "/%s//%s/%s" % (molecule, chain, resi)
cmd.get_wizard().do_select(selection)
cmd.frame(str(mutframe))
cmd.get_wizard().apply()
# cmd.set_wizard("done")
cmd.set_wizard()
# cmd.refresh()
cmd.extend("mutate", mutate)
或者直接下载笔者编写的脚本 mutate.py
在 PyMOL 命令行,运行下述命令,就可以添加mutate命令了。
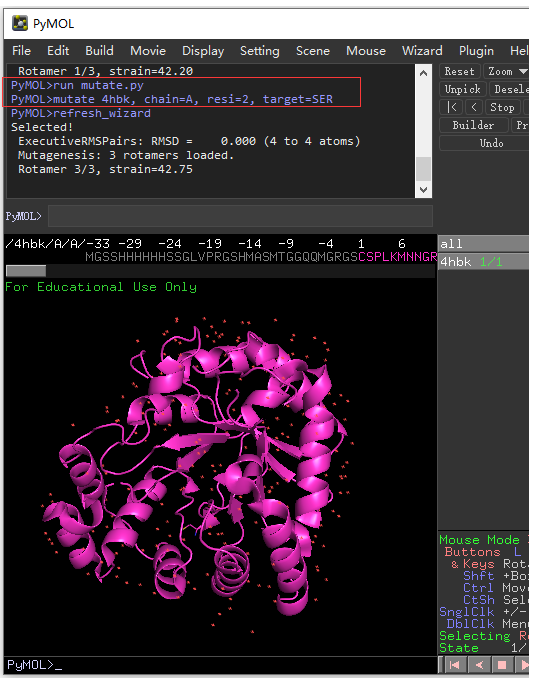
PyMOL>run mutate.py
PyMOL>mutate 4hbk, chain=A, resi=2, target=SER
单点全扫描突变插件¶
插件:
from pymol import cmd
from pymol import cmd
#refer: https://pymolwiki.org/index.php/Rotkit
#qq group: 294266356
def mutate(molecule, chain, resi, target="CYS", mutframe="1"):
target = target.upper()
cmd.wizard("mutagenesis")
cmd.do("refresh_wizard")
cmd.get_wizard().set_mode("%s" % target)
selection = "/%s//%s/%s" % (molecule, chain, resi)
cmd.get_wizard().do_select(selection)
cmd.frame(str(mutframe))
cmd.get_wizard().apply()
# cmd.set_wizard("done")
cmd.set_wizard()
# cmd.refresh()
cmd.extend("mutate", mutate)
resns=['']
def mutate_others(objname="P4G3",chain='A',resi=1):
'''
help: mutate_others objname,118
mutate_others P4G3,A,7
'''
RESIDUES=['ALA', 'ILE', 'LEU', 'MET', 'VAL', 'PHE', 'TRP', 'TYR', 'ASN', 'CYS', 'GLN', 'SER', 'THR', 'ASP', 'GLU', 'ARG', 'HIS', 'LYS', 'GLY', 'PRO']
for resn in RESIDUES:
newfile="%s_%s_%s"%(objname,resi,resn)
# cmd.mutate()
cmd.do( 'mutate %s,%s,%s,%s'%(objname, chain, resi, resn))
cmd.save("%s.pdb"%newfile,objname)
cmd.extend("mutate_others", mutate_others)
用法:
对P4G3蛋白的A链上的7号残基进行全扫描突变并保存文件
mutate_others P4G3,A,7
平均构象 states_avg.py¶
Cameron Mura <cmura@ucsd.edu>在2005年写了一个脚本 average3D.py ,
该脚本太旧而无法使用,笔者在其基础上进行了修改,创建脚本 mutate.py 。
注解
在修改过程,遇到内存崩溃的坑,解决办法为states创建独立的变量。 obj_states=[]
代码如下:
"""
AUTHOR:
Cameron Mura <cmura@ucsd.edu>; 07/2005
update: zhaoqiang Chen <744891290@qq.com>; 05/2022 support python3
http://www.bioinformatics.org/cgi-bin/viewvc.cgi/pdbwiki/trunk/scripts/average3d/average3d.py?revision=36&view=markup
SYNOPSIS:
Python module "average3d.py" for averaging 3D coordinates of multiple states
of a single PyMOL molecular object
USAGE:
See this source file for now... in particular, any notes under individual
function definitions, such as 'avgStates()'.
refer: average3D.py
http://www.bioinformatics.org/cgi-bin/viewvc.cgi/pdbwiki/trunk/scripts/average3d/average3d.py?revision=36&view=markup
The script(average3D.py) is too old and outdated to use, so I created the script "states_avg.py".
run e:/states_avg.py
fetch 1wym
states_avg 1wym
"""
from pymol import cmd
from math import sqrt,pow
def states_avg(objectstr='all'):
##save mem
# cmd.undo_disable()
objectstr= str(objectstr)
first=int(1)
last=int(0)
num_states_tot = cmd.count_states(objectstr)
print("num states",num_states_tot)
last=num_states_tot
num_states2avg = last - first + 1
newobj='%s_avg'%(objectstr)
cmd.create(newobj,objectstr,1)
tmpobj='%s_avgtmp'%(objectstr)
cmd.create(tmpobj,objectstr,1)
newobj_chempy = cmd.get_model(tmpobj,1)
obj_states=[]
for s in range(1,num_states2avg+1):
stat = cmd.get_model(objectstr,s)
obj_states.append(stat)
# any object index start from 1
for at in newobj_chempy.atom:
this_at_idx = at.index
sum_x = sum_y = sum_z = 0.0
avg_x = avg_y = avg_z = 0.0
for s in range(1,num_states2avg+1):
this_x = obj_states[s-1].atom[this_at_idx-1].coord[0]
this_y = obj_states[s-1].atom[this_at_idx-1].coord[1]
this_z = obj_states[s-1].atom[this_at_idx-1].coord[2]
sum_x += this_x
sum_y += this_y
sum_z += this_z
# print(sum_x,sum_y,sum_z)
avg_x = sum_x / num_states2avg
avg_y = sum_y / num_states2avg
avg_z = sum_z / num_states2avg
# # at.coord[0] = avg_x
# at.coord[0] = avg_y
# at.coord[0] = avg_z
# print(avg_x,avg_y,avg_z,this_at_idx)
cmd.alter_state(1,'%s and index %d'%(newobj,this_at_idx),'x=%.3f'%avg_x)
cmd.alter_state(1,'%s and index %d'%(newobj,this_at_idx),'y=%.3f'%avg_y)
cmd.alter_state(1,'%s and index %d'%(newobj,this_at_idx),'z=%.3f'%avg_z)
# print("update success")
print("update success ^ ^ congratulations")
cmd.delete(tmpobj)
# make it directly callable in PyMOL:
cmd.extend('states_avg',states_avg)
使用方法:
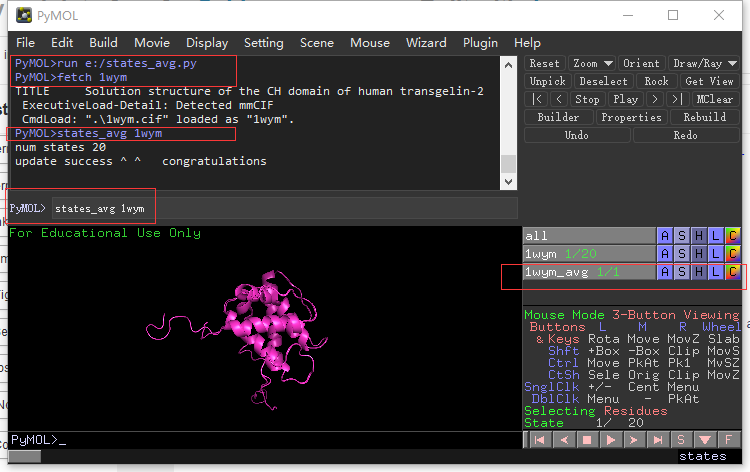
下载笔者编写的脚本 mutate.py ,保存到E盘,
这里以PDB ID: 1WYM为例,运行如下命令:
run e:/states_avg.py
fetch 1wym
states_avg 1wym
PyMOL 拓展命令 spectrum_states¶
功能:基于state进行着色 地址:https://pymolwiki.org/index.php/Spectrum_states 下载:https://raw.githubusercontent.com/Pymol-Scripts/Pymol-script-repo/master/spectrum_states.py
用法:
这里以对名C9的object着色为例,
run d:/spectrum_states.py
as sphere,C9
spectrum_states C9, sphere, red pink
注解
设置object的表现形式,这一步非常重要,不能省略。
PyMol的宏录制¶
点击 File->Log File-> Open File 填写宏的名字 xxx.pml 然后操作PyMOL, 操作完成后点击 File->Log File-> close; 文本编辑器(如vscode)打开宏文件就可以查看里面的命令。
PyMol插件PyVOL推荐¶
PyVOL介绍¶
pyvol github 仓库: https://github.com/schlessinger-lab/pyvol 。 PyVOL是一个对蛋白口袋进行可视化的pymol插件。
口袋通常由氨基酸残基及其包裹的空间组成,为了可视化的美观,建议在空间中填充格点。
PyVOL安装¶
经过我的测试,发现无法在windows上的pymol2+上面进行安装。 只能安装在linux的pymol上。
通过conda安装msms
conda install -c bioconda msms
安装后,把msms所在目录添加到环境变量path中。
从官网下载 pyvol插件 pyvol-installer.zip:
如果如法下载,可通过百度云进行下载:
链接:https://pan.baidu.com/s/1TfYZDXBgZPUZxFG2S58H0Q
提取码:j9z7
在pymol中点击 Plugins -》 Plugin Manager -》 Install New Plugin -》 Install from local file -》 Choose file… 进行安装。
pyvol使用¶
注解
对于一些冷冻电镜解析的超大蛋白,使用该软件可能会报错。
建议参数如下:
Protein Pymol Selection: all
minimum volume: 100
dsplay mode: Sphere
基于产生的结果,对口袋进行修饰(删除冗余格点),并创建新的object,显示成surfce形式。

PyMol插件parKVFinder-win推荐¶
ParKVFinder-win介绍¶
ParKVFinder github 仓库: https://github.com/LBC-LNBio/parKVFinder-win 。 ParKVFinder 是一个对蛋白口袋进行可视化的pymol插件。
注解
需要使用schrodinger版本的pymol
口袋通常由氨基酸残基及其包裹的空间组成,为了可视化的美观,建议在空间中填充格点。
ParKVFinder-win安装¶
这是专门针对windows系统的软件, PyMOL2+的插件。
安装流程如下:
- step1 通过github仓库下载 parKVFinder-win-master.zip 文件。
- step2 解压parKVFinder-win-master.zip到本地路径,比如解压到D盘目录,在环境变量中增加变量”KVFinder_PATH”,值设置为”D:parKVFinder-win-master”或者”D:parKVFinder-win-masterparKVFinder-win64.exe”。在个人电脑上设置为前者显示成功。
- step3 找到PyMOL的安装目录,打开里面的**Conda-Prompt.bat**的命令窗口,输入命令”pip3 install toml”,安装python模块toml。
- step4 从github的 release仓库 中下载最新版本的 PyMOL2-parKVFinder-Tools.zip 的PyMOL插件。
- step5 打开pymol2,点击 plugin->plugin manager -> install new plugin ->choose file ->PyMOL2-parKVFinder-Tools.zip。
看到提示“Plugin pymol2-parKVFinder-Tools has been installed” 说明安装成功。
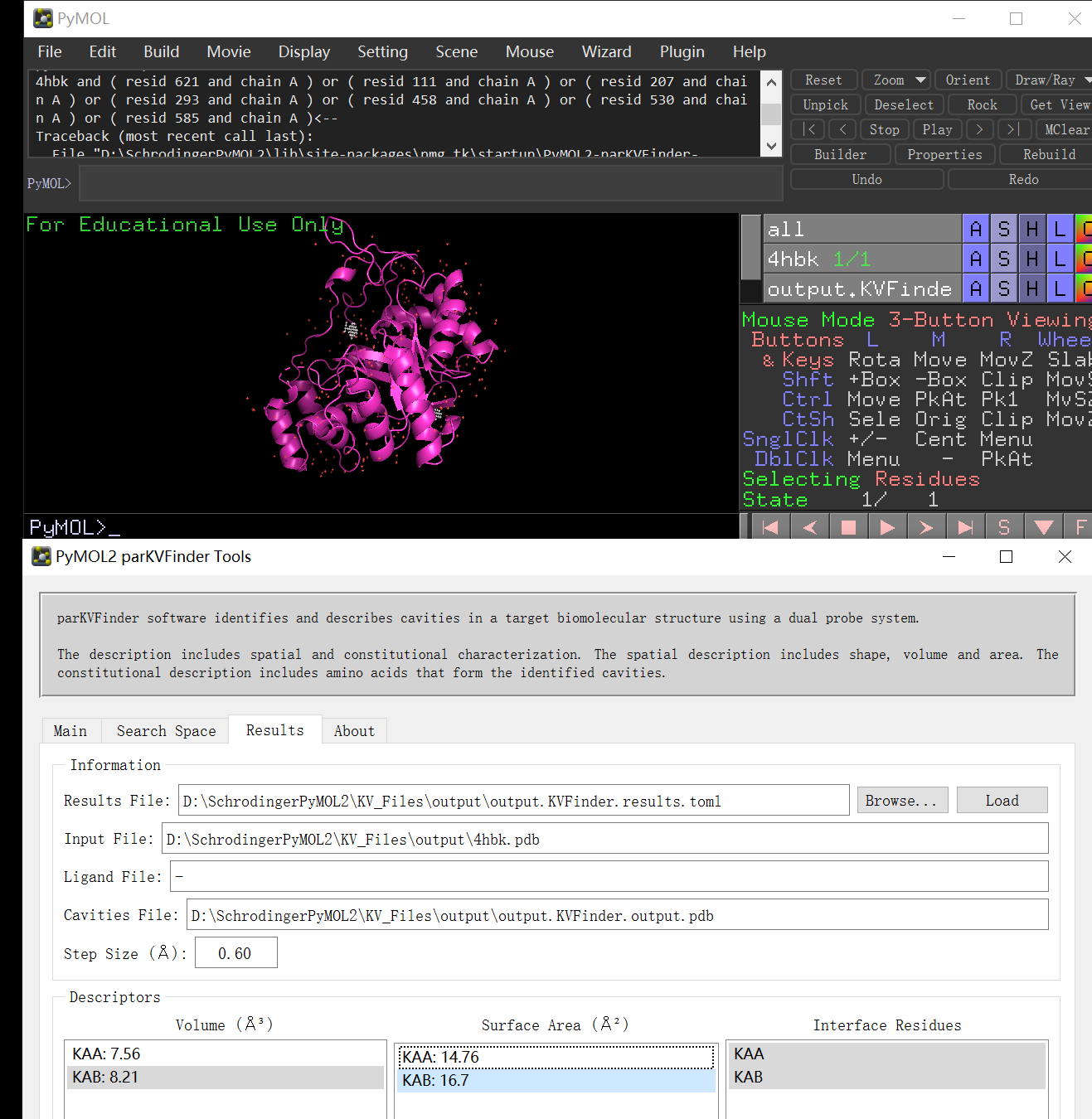
ParKVFinder-win使用¶
载入蛋白,点击plugin->pymol2-parKVFinder-Tools,打开相应的界面。 点击 refresh->run kvfinder就可以了。 结果如下,可以看到检测到的空腔的体积和表面积以及氨基酸残基组成。
PyMol插件CAVER推荐¶
CAVER介绍¶
CAVER 官网: https://caver.cz/index.php
百度网盘链接: https://pan.baidu.com/s/1QWsPAvuyVV3M1k22jI_M7w?pwd=nihg
CAVER 是一款用于分析蛋白质通道和孔道的工具,特别适用于可视化分子在蛋白质结构中的解离路径。 隧道是从埋在蛋白质核心中的空腔通向周围溶剂的空隙通道。 通道穿过蛋白质结构,其两端都通向周围溶剂。
注解
CAVER 提供了多种版本,方便用户根据需求选择,包括 1. 命令行版本、 2. PyMol 插件以及 3. 图形界面版本(CAVER Analyst) 4. 在线服务器版本: https://loschmidt.chemi.muni.cz/caverweb/
主要功能:
- 对动力学轨迹进行可视化;比如可视化SMD过程中小分子的解离路径;
这里我主要介绍Caver的PyMOL插件版本。
Caver插件安装¶
这是专门针对windows系统的软件, PyMOL2+的插件。
安装流程如下:
- step1 通过github仓库下载
caver-pymol-plugin-3.0.3.zip 文件。
- step2 安装JRE8,并检查JRE版本。
(base) C:\Users\Lenovo>java -version
java version "1.8.0_411"
Java(TM) SE Runtime Environment (build 1.8.0_411-b09)
Java HotSpot(TM) 64-Bit Server VM (build 25.411-b09, mixed mode)
电脑上安装的是 Java SE Runtime Environment(JRE)版本 1.8.0_411。 这是 Java 8 的一个更新版本,其中:
1.8.0_411 表示这是 Java 8 的第 411 个更新版本。
Java(TM) SE Runtime Environment 表示这是 Java 的标准版运行时环境。
- step5 打开pymol2,点击 plugin->plugin manager -> install new plugin ->choose file ->caver-pymol-plugin-3.0.3.zip 。
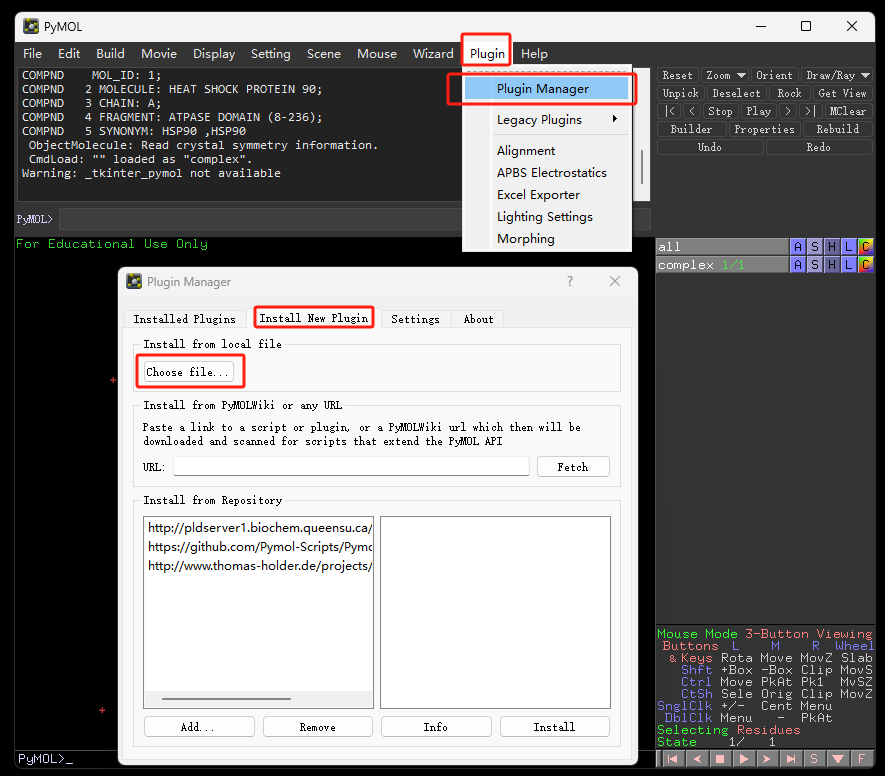
看到提示“Plugin CAVER3 has been installed” 说明安装成功。
CAVER3的使用¶
载入T4 lysozyme蛋白(PDB ID: 4W52),点击plugin->Legacy Plugins>Caver 3.0.03 ,打开相应的界面。
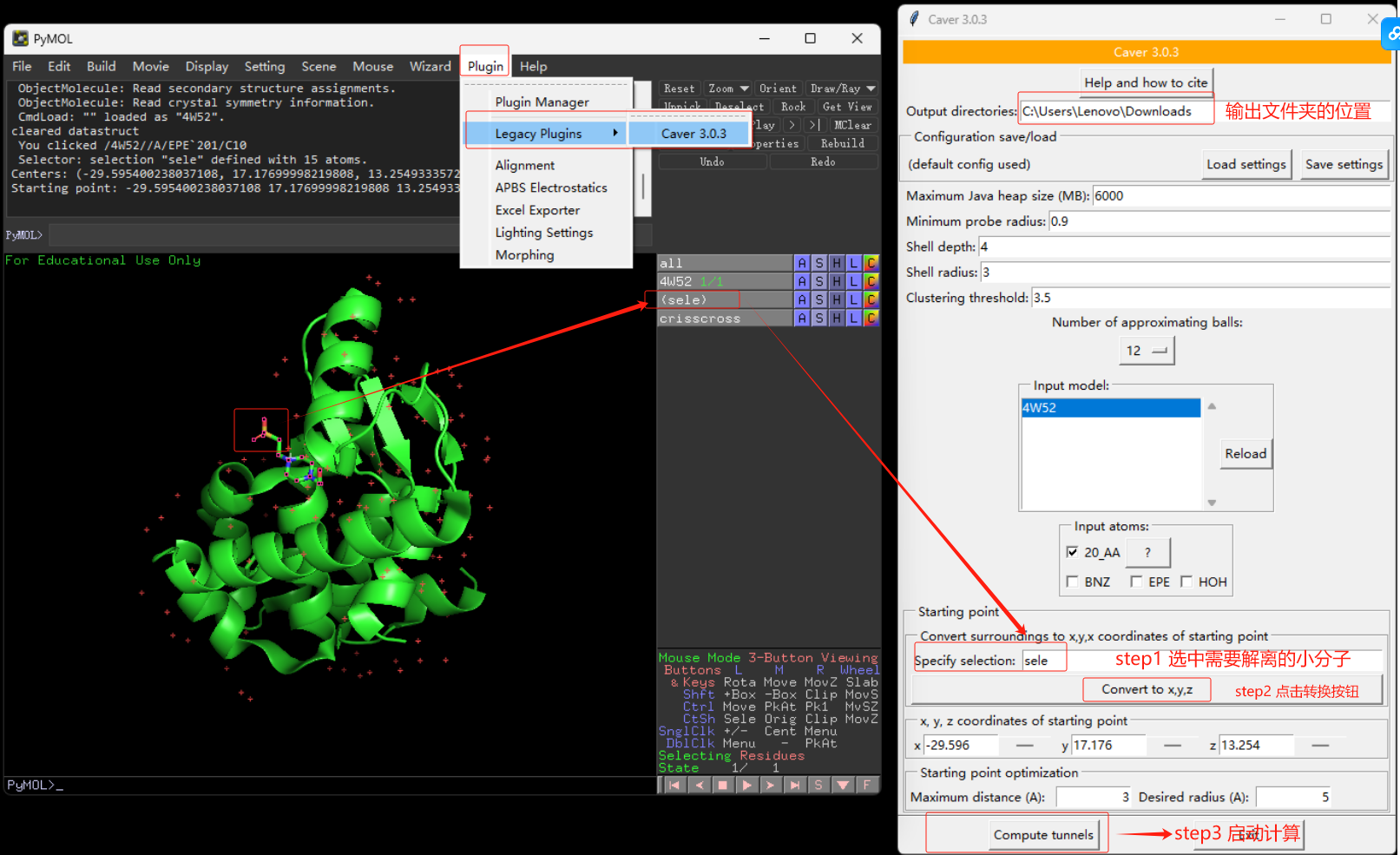
一共找到了2条通道,
和 Caver WEB: https://loschmidt.chemi.muni.cz/caverweb/?action=results&job=gpmkqm&analysis=single_structure& 结果一致,如下图所示: