FAQs¶
Q1. 蛋白断裂¶
Q1. 老师您好 我用pymol显示相互作用的时候 show sticks 然后蛋白的beta折叠就断了 咋样显示让蛋白更好看一些不断裂呢?
A1. 参见 入门教程中的 show 和 show as 的区别。
Q2. 界面暗淡¶
Q2. pymol里面打开结构很暗,重装软件也没有用,怎么解决?
A2. OpenGL模块不兼容,在setting中rendering中设置关闭opengl,或者重新装opengl驱动。或者点击 File-> reinitialize->original settings.
Q3. 消除虚线¶
Q3. 请问pymol如何消除自动补全的虚线啊?
A3. 氨基酸缺失或者一段seq离开了自己原来的位置,pymol 会在它认为链接的地方加虚线。如果想要掩藏虚线,将其拆分成2个独立的object就可以了。
Q4. 透明物体消失¶
Q4. ray 渲染后内部非透明物体消失?
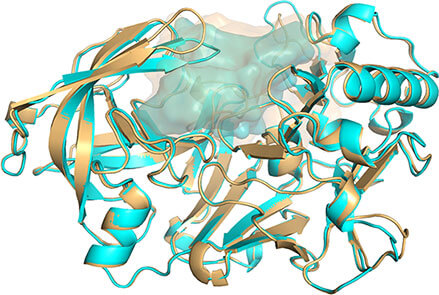
Fig 1. befor ray |
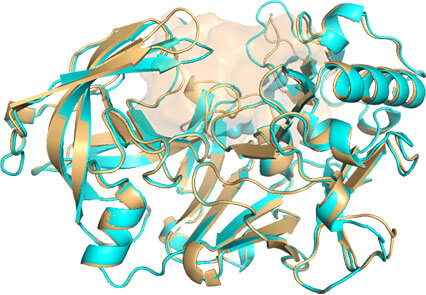
Fig 2. after ray |
可通过间接的方法实现ray后内部非透明物体可见,保留3张图片,然后1.openPOCKET; 2. closePocket; 3.proteintCartoon. 然后把3张图图片导入到PhotoShop中,从上到下一次是123的顺序就可以了。

Fig 1. openPocket |
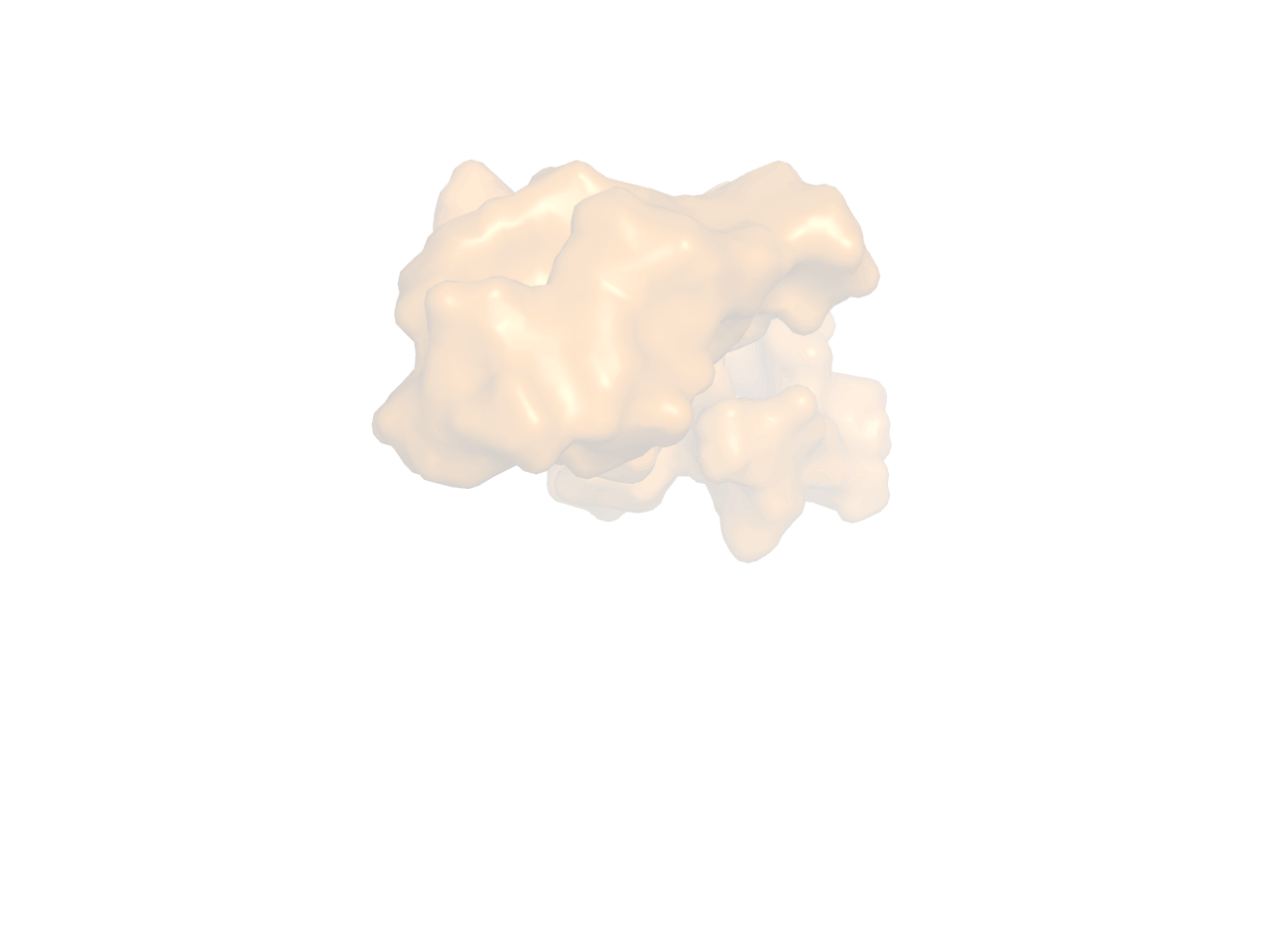
Fig 2. closePocket |
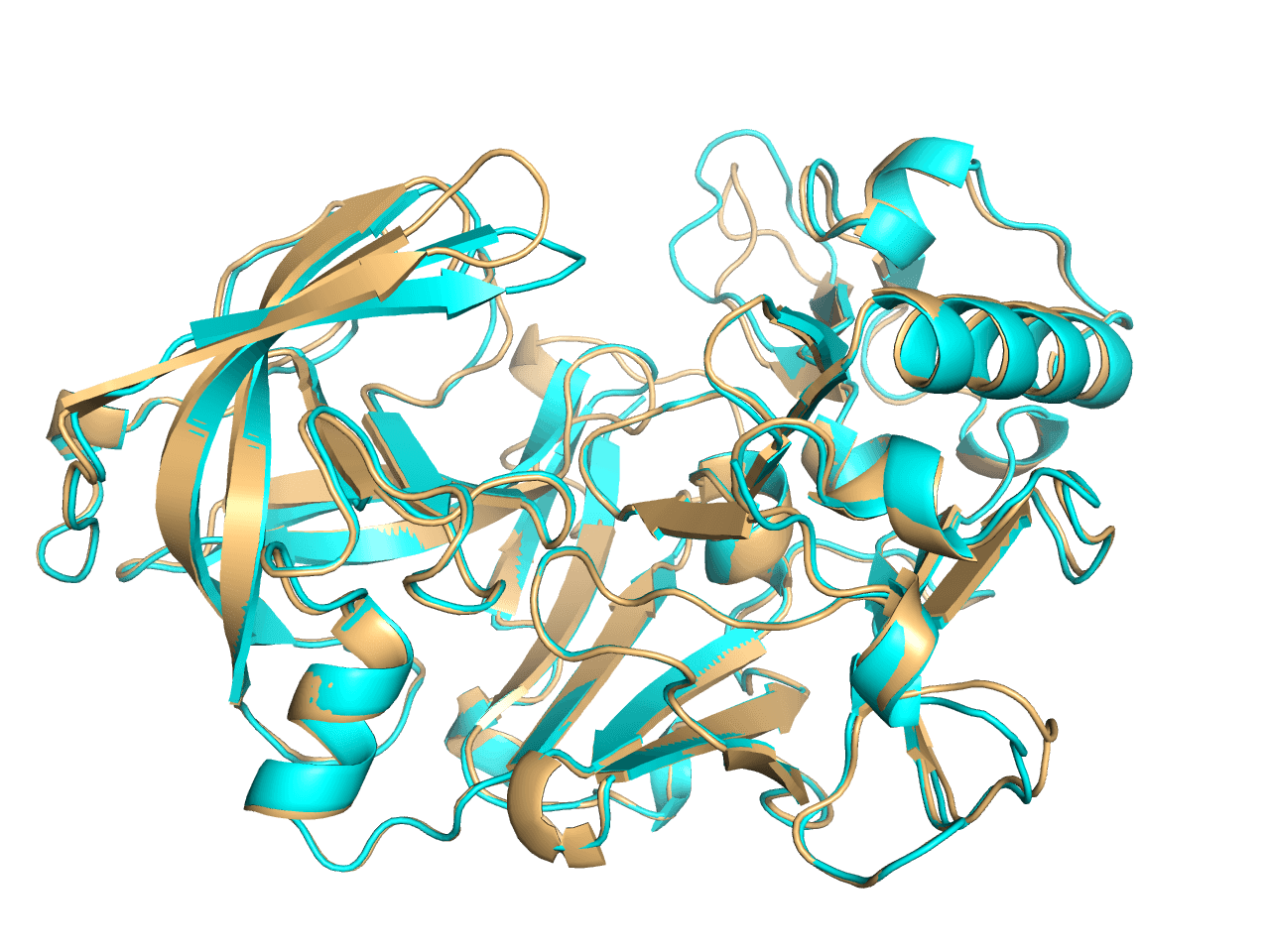
Fig 3. proteinCartoon |
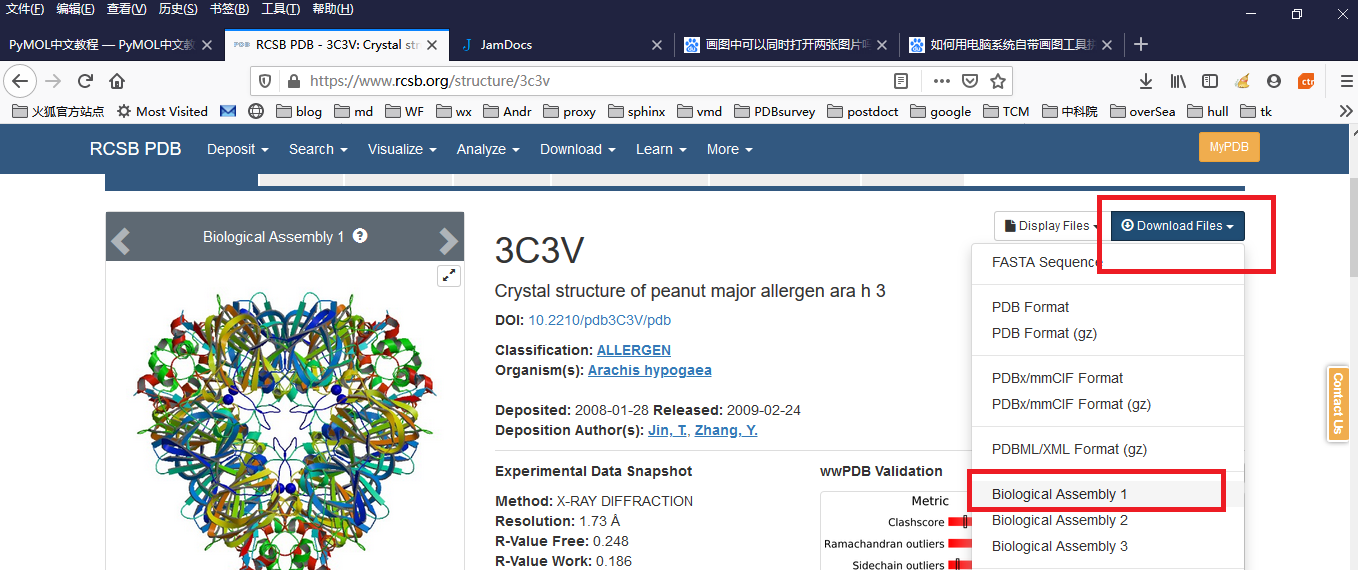
Q5. Biological Assembly¶
Q5. 想请教老师们一个问题。我在pdb官网想下载一个蛋白,这个蛋白应该是个六聚体,图上也是这样显示的(功能上是多聚体), 但是我在下载栏下载pdb format下载结果却是单体,请问老师们知不知道这个问题如何解决?

A5. 方法一 直接在PDB网站上下载相应的生物多聚体,点击Dowoload Files 然后选择对应的Biological Assembly. 在PYmol 中打开该文件,点击 Action->state->split 就可以了。适合PyMOL2。
方法二 直接打开单体文件,点击Action->generate->symmetry mates->within 5a (合适的半径进行拓展) 然后删除冗余的单体就可以了。
Q6 三角形表面¶
Q6. PyMOL如何渲染出下面图片的效果(三角形表面)?
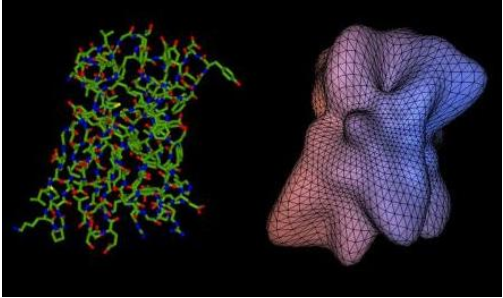
- A6. 从图中我们可以看出是光滑表面上加了一层三角形网格。该图片来源 Hex 网站 。
- 具体操作步骤,在pymol中第一次渲染普通表面,然后ray 背景透明保存成第一张图片。 然后在设置表面为三角形,
set surface_type,2
然后ray,然后再渲染表面,背景透明保存成第二张图片,注意该操作对电脑要求较高,建议使用20个以内的氨基酸蛋白尝试。 最后再在photohop中,把两张图片叠合在一起即可。
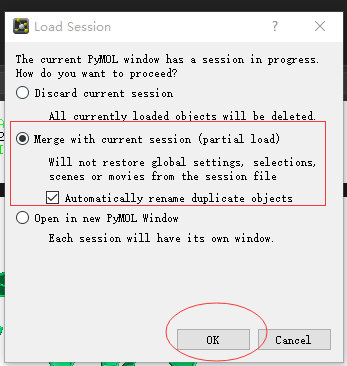
Q7. 如何同时打开两个pse文件?¶
A7. 首先File->open 打开第一个pse文件,然后继续File-open 选择第二个pse文件, 会弹出对话框,有3个选项:
- 打开最新的pse,原来的pse丢弃;
- 同时打开两个pse,如果有object重名,则自动修改名字
- 在新的窗口中打开新的pse.
因此,选第二个选项就可以打开2个pse 文件了。
Q8. 疏水氨基酸和亲水氨基¶
如何在pymol里面可以鲜明地显示这种亲水氨基酸残基和疏水氨基酸残基区域吗,如下图所示?
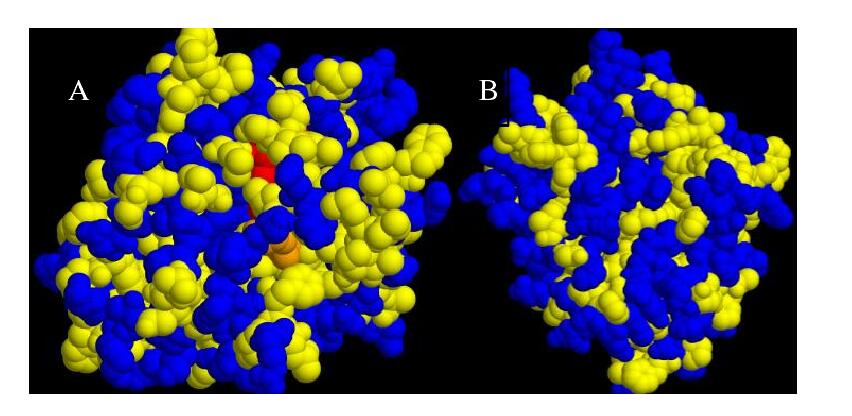
A8. 疏水氨基酸有9个:Gly,Ala,Val,Leu,Ile,Pro,Phe,Met,Trp。 The nine amino acids that have hydrophobic side chains are glycine (Gly), alanine (Ala), valine (Val), leucine (Leu), isoleucine (Ile), proline (Pro), phenylalanine (Phe), methionine (Met), and tryptophan (Trp).
非电极性(亲水)氨基酸有6个:Ser,Thr,Cys,Asn,Gln,Tyr。
注解
氨基酸性质的划分是基于其侧链R基团进行划分的。
Amino acids are grouped according to what their side chains are like.
The nine amino acids that have hydrophobic side chains are glycine (Gly), alanine (Ala), valine (Val),
leucine (Leu), isoleucine (Ile), proline (Pro), phenylalanine (Phe),
methionine (Met), and tryptophan (Trp).
Six amino acids have side chains that are polar but not charged.
These are serine (Ser), threonine (Thr), cysteine (Cys), asparagine (Asn), glutamine (Gln), and tyrosine (Tyr).
These amino acids are usually found at the surface of proteins, as discussed in the Proteins 2 module.
在命令行窗口,输入下面4个命令就可以。
color yellow, resn Gly+Ala+Val+Leu+Ile+Pro+Phe+Met+Trp
as sphere, resn Gly+Ala+Val+Leu+Ile+Pro+Phe+Met+Trp
as sphere, resn Ser+Thr+Cys+Asn+Gln+Tyr+Asp+Glu+Arg+Lys+His
color blue, resn Ser+Thr+Cys+Asn+Gln+Tyr+Asp+Glu+Arg+Lys+His
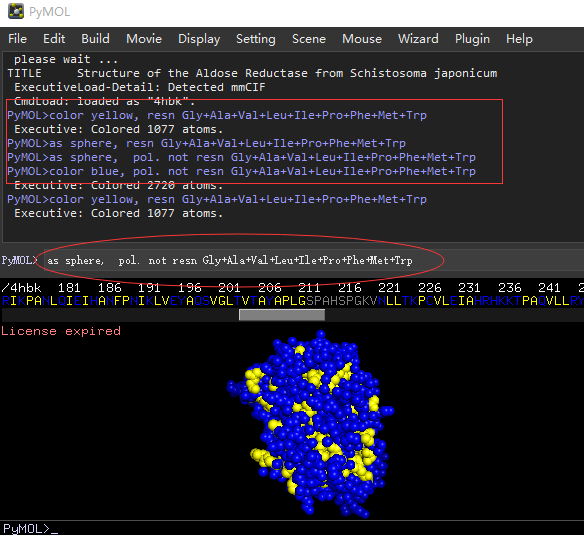
Q9. 缺失残基resi 26-39怎么在PyMOL中用虚线表示?¶
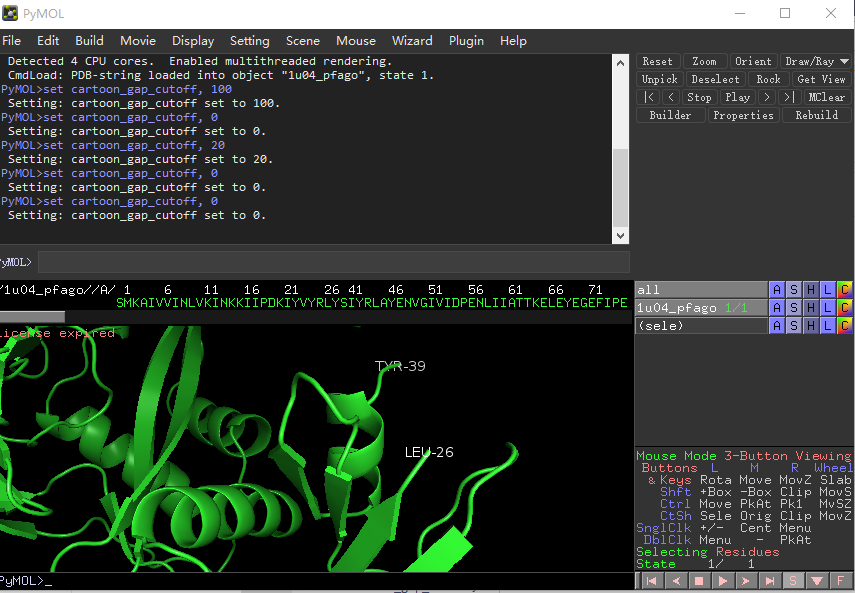
缺失残基resi 26-39怎么在PyMOL中用虚线表示? 如果缺失残基的数目小于10个,在pymol 1.8.2+自动在gap处添加虚线。 如果超过10个,则需要手动设置是否显示虚线。 A9. 关闭虚线
set cartoon_gap_cutoff, 0
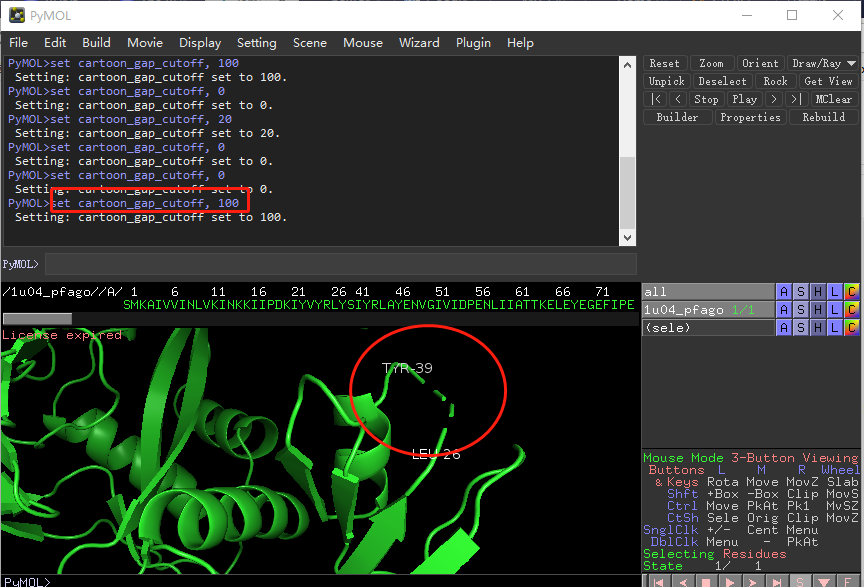
设置阈值,如果缺失的残基小于阈值,则显示虚线,默认是10
set cartoon_gap_cutoff, 100
Q10. 不能正确显示cartoon 等二级结构¶
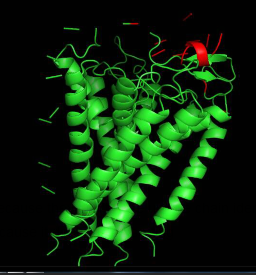
分析后发现是残基的链接顺序和编号顺序不一致导致的,解决办法由有2种。 22号和23号在PyMOL默认认为相连号码的残基在空间是相连的。
方法一: 修改pdb文件,调整编号顺序。 方法二: 自动识别残基的可能链接顺序,暂时不支持rebuild导出正确的顺序。
set retain_order,1
dss all
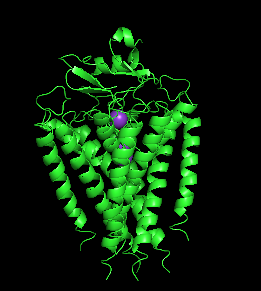
修改后的效果
Q11. 如何自定义label¶
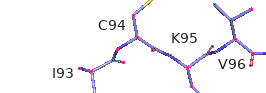
如下图所示,显示了氨基酸的单字符和字符的位置ID,我如何指定将I93 指只显示为I,而不显示93呢? 请问有谁知道该如何处理吗?谢谢
通过label命令进行自定义就可以了,Label命令格式如下
label atom, customLabel
第一个参数: 定位需要label的原子。 第二个参数: 字符串
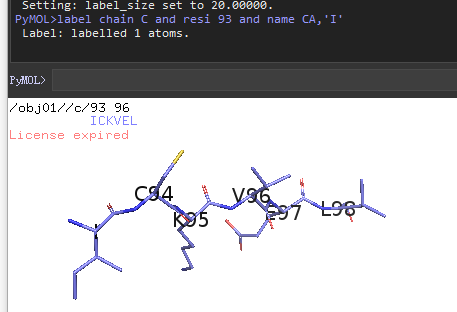
在PyMOL中运行下述命令 label chain C and resi 93 and name CA,’I’ 就可以了。
Q12. 添加、修改chain ID¶
我模拟出来的PDB文件没有链的ID号,我想自己编辑一个链ID号上去,请问怎么进行编辑
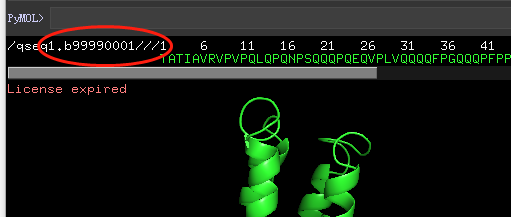
方法一: 在pymol 命令窗口输入命令
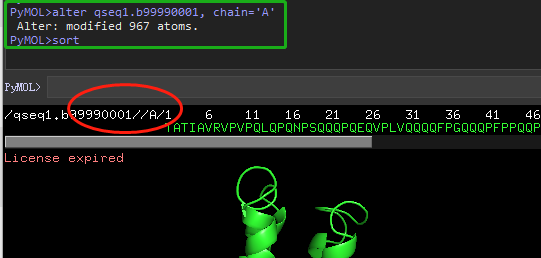
PyMOL>alter qseq1.b99990001, chain='A'
Alter: modified 967 atoms.
PyMOL>sort
方法二: 在文本中修改
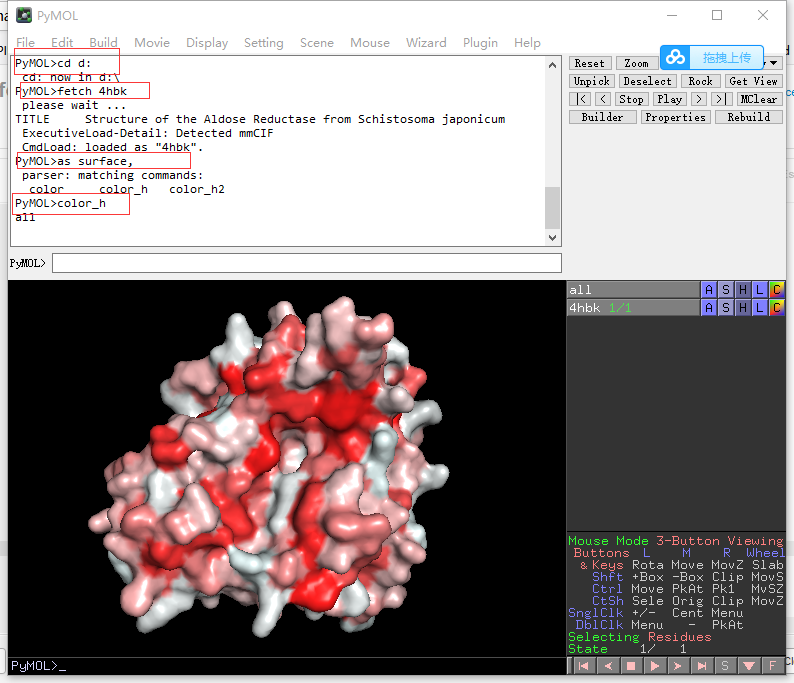
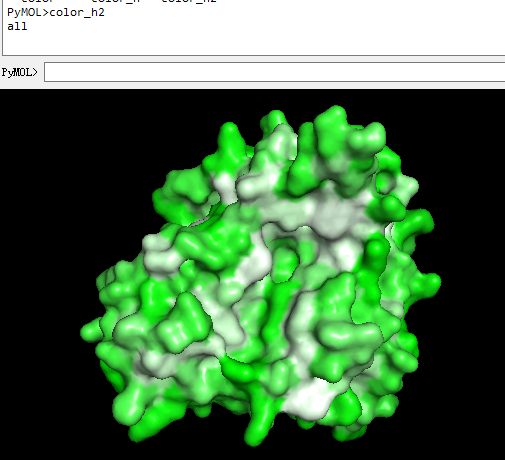
Q13. 用pymol怎么显示蛋白的亲水疏水表面¶
下载脚本 color_h 。 到工作目录下面(如D:/),打开pymol切换到工作目录,载入蛋白,运行脚本。 color_h 绘制疏水表面,红色越深疏水越强; color_h2 绘制亲水表面,绿色越深,亲水越强。
运行 color_h 命令,
运行 color_h2 命令,
Q14. 颜色按钮无法正常显示颜色¶
方案一: 尝试对object着一个颜色,然后再点击颜色按钮;
方案二: 使用命令行,尝试不同的颜色编码
util.cba(1,"obj01",_self=cmd)
util.cba(2,"obj01",_self=cmd)
util.cba(3,"obj01",_self=cmd)
util.cba(4,"obj01",_self=cmd)
util.cba(5,"obj01",_self=cmd)
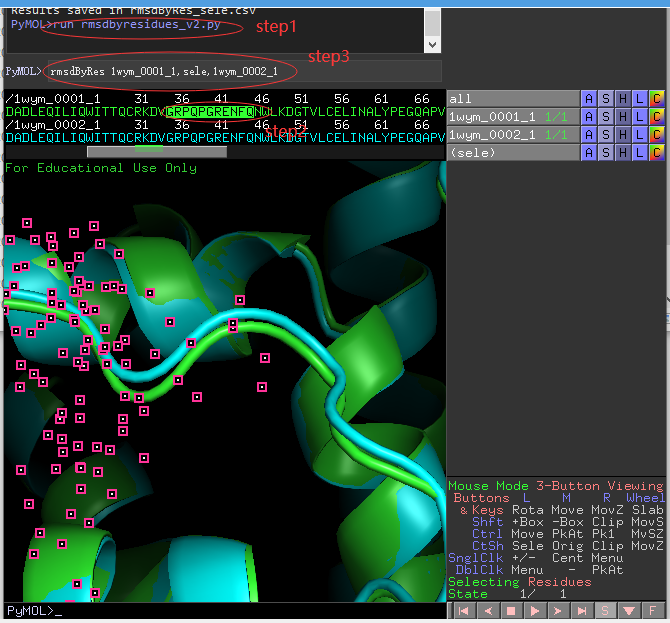
Q15. 如何RmsdByResidue插件?¶
这个脚本怎么使用呀? https://pymolwiki.org/index.php/RmsdByResidue
第一个问题我装不上,我看了一下是不是因为他是根据python2 写的?
第二个 我有的同学装上了,但是也不会用。
我打开发现这个脚本是我师兄Zhenting Gao写的,可能使用频率不是很高,没有维护下去。 里面的细节处理值得学习一下。
第一个问题主要有2个问题:
- 上传到pymolwiki后缩进不正确,建议提高文件下载链接;
- 所有的print添加括号,这样可以兼容python2 和python3。
可直接下载笔者修改后的脚本 rmsdbyresidues_v2
第二个问题如何使用,
`
rmsdByRes referenceProteinChain,sel, targetProteinChain
`
注解
- 使用该命令前,先对结构进行align叠合。
- 对应氨基酸的原子数目要一致,不能一个有氢原子,另一个没有氢原子。建议删除氢原子。
用法如下图所示:
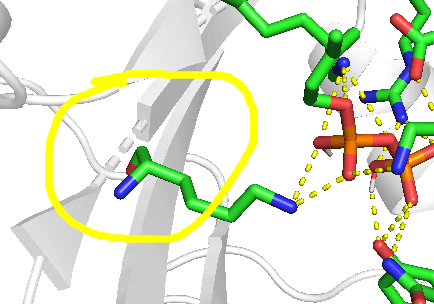
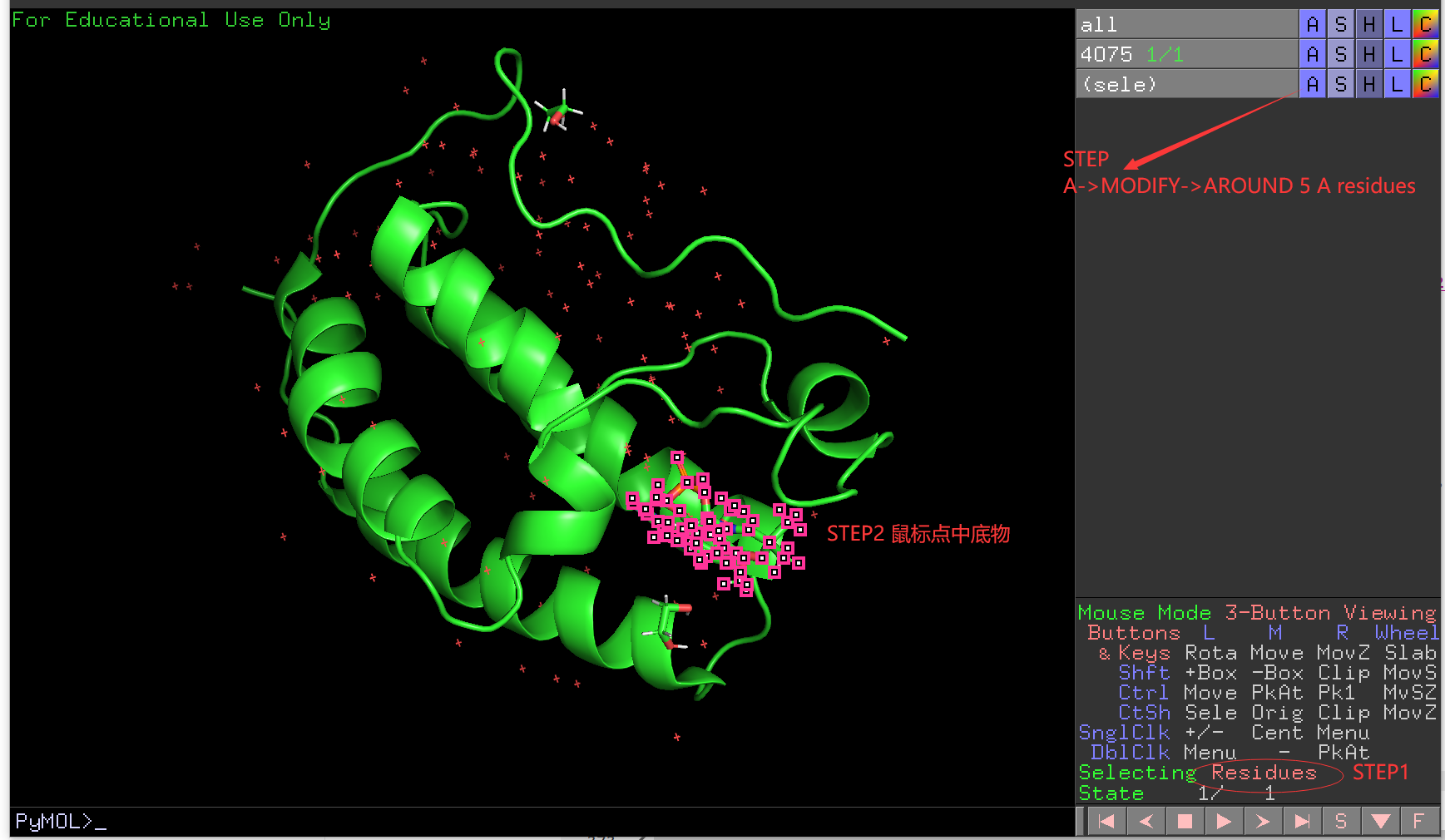
Q16. 如何找底物附近的氨基酸?¶
以PDB ID: 4O75为例,在mode为residues模式先,选中底物,出现sele,
在sele的对象上点击A->MODIFY->RESIDUES WITHIN 4A; 然后点击A->copy to object new。
创建了object1, 对object1进行show as sticks和label residues就可以看到附近4A的残基了。
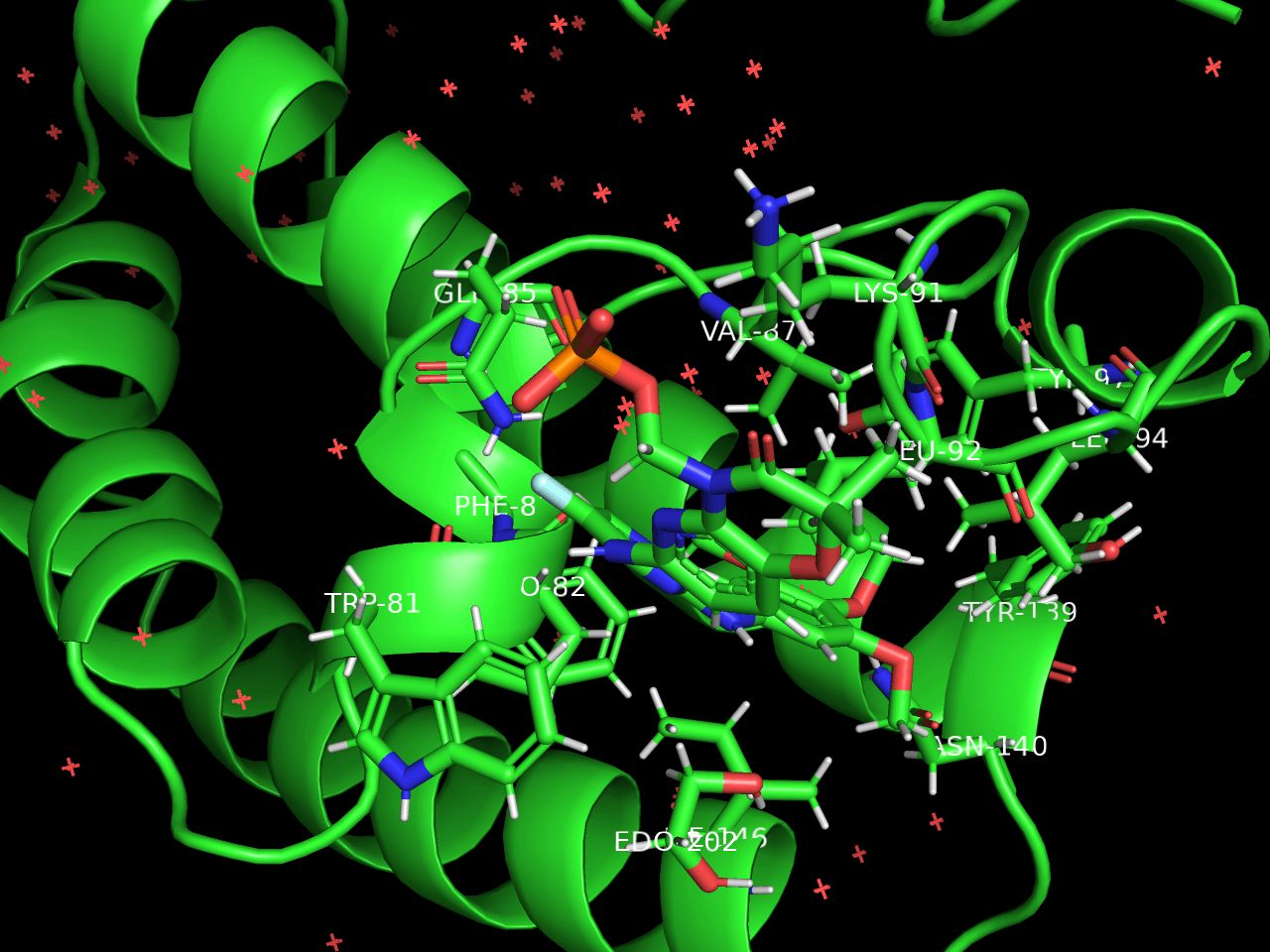
Q19. pymol有没有命令返回在氨基酸编号,在小分子4A范围内氨基酸的编号?¶
这里以JAK1 (PDB ID:3EYG)为例,
示例代码如下:
from pymol import cmd
select mt1around5, name ca and byres (resn MI1 around 5 and polymer) and name ca
atoms =cmd.get_model("mt1around5").atom
[ print(a.resn,a.resi) for a in atoms]
Q20. 请问一个nmr的pdb文件中有很多个构象,如何求他的平均结构呢?¶
下载笔者编写的脚本 mutate.py ,保存到E盘,
这里以PDB ID: 1WYM为例,运行如下命令:
run e:/states_avg.py
fetch 1wym
states_avg 1wym
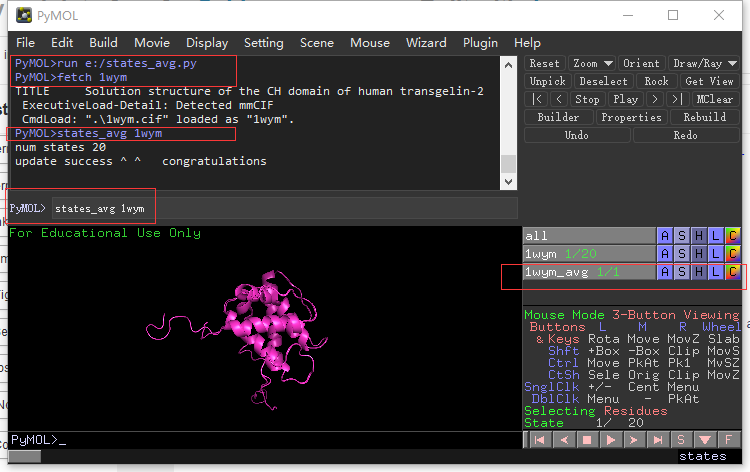
关于该脚本的更详细说明,参见 高级教程->PyMol脚本->平均构象 。
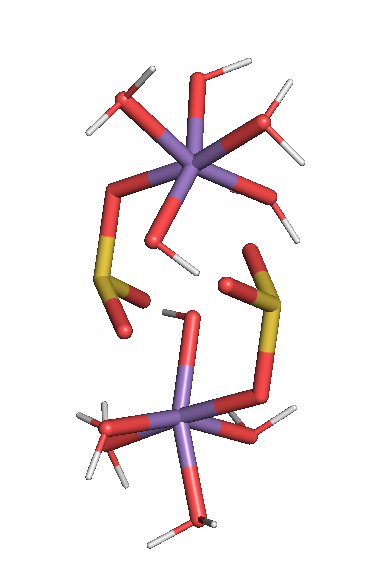
Q22. 让含氢原子的键的stick和其他键的粗细保持一致?¶
默认pymol中含有氢原子的键的stick模式,要比其它键更细,这有什么办法可以让键的粗细保持一致?
命令如下:
PyMOL>set_bond line_width, 10,obj01
Setting: line_width set for 36 bonds in object "obj01".
PyMOL>as lines, obj01
PyMOL>ray
默认效果:
修改后效果:
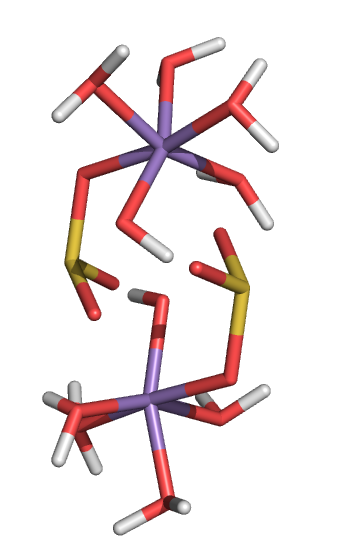
注意事项:
注解
- 只有在ray的状态下可以看到效果,点一下就消失。
- 存图的时候,进行ray。
Q23. PyMOL工作路径异常怎么恢复?¶
场景: 某某修改PyMOL的环境变量PATH后,发现工作路径异常, 每次打开pdb文件或者sdf文件,工作路径自动定位到C盘。
需求:每次打开pdb文件,工作路径自动切换到pdb文件所在路径。
测试发现和环境变量无关,暂时不清楚是什么原因导致工作路径异常。
File -> Reinitialize-> Oiginal Settings 进行设置。 如果一次不行,可以多重复几次。
Q24. 如何在heme中心的Fe在垂直上方加一个O原子,形成Fe=O,键长约1.64埃?¶
以PDB ID: 3CBD 为例,如下图所示,我们可以看到HEME下方有一个Cys400和Fe进行配位。
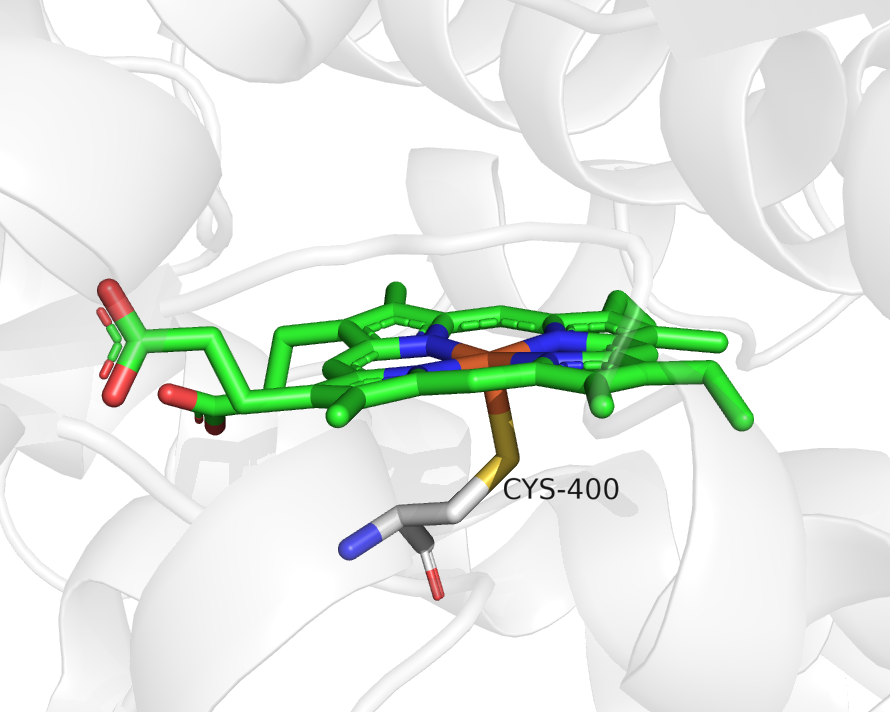
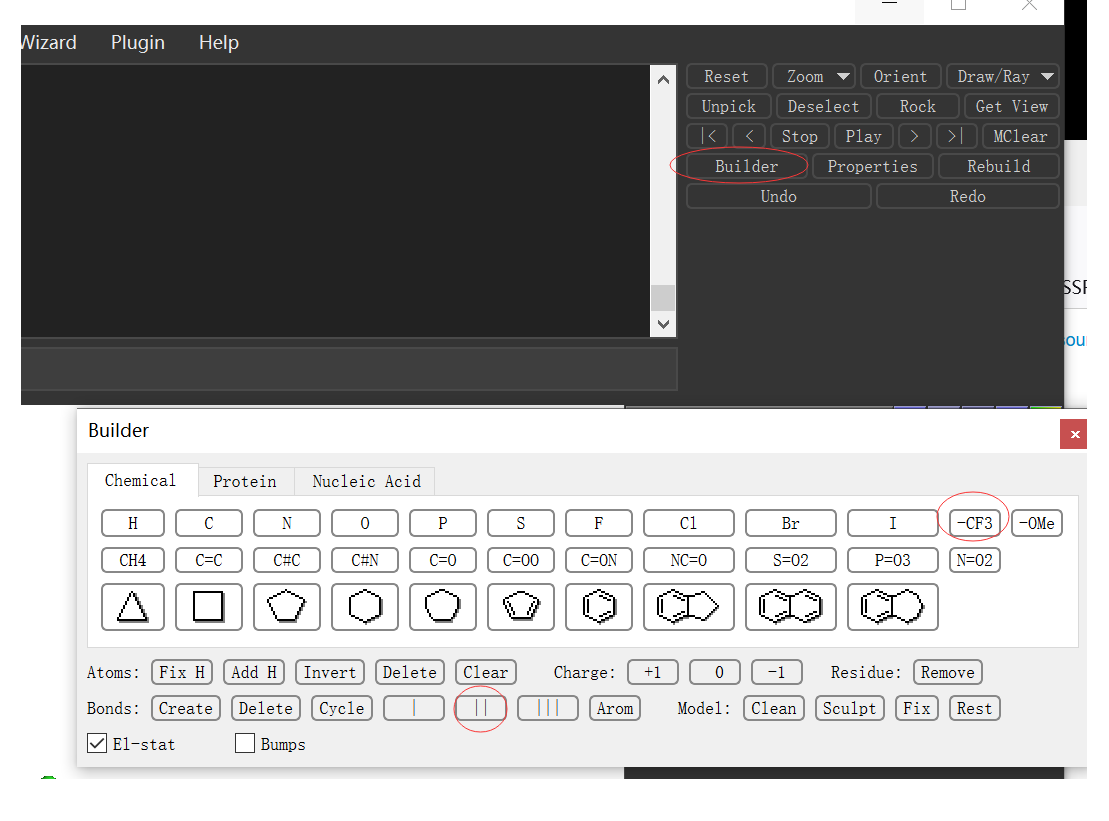
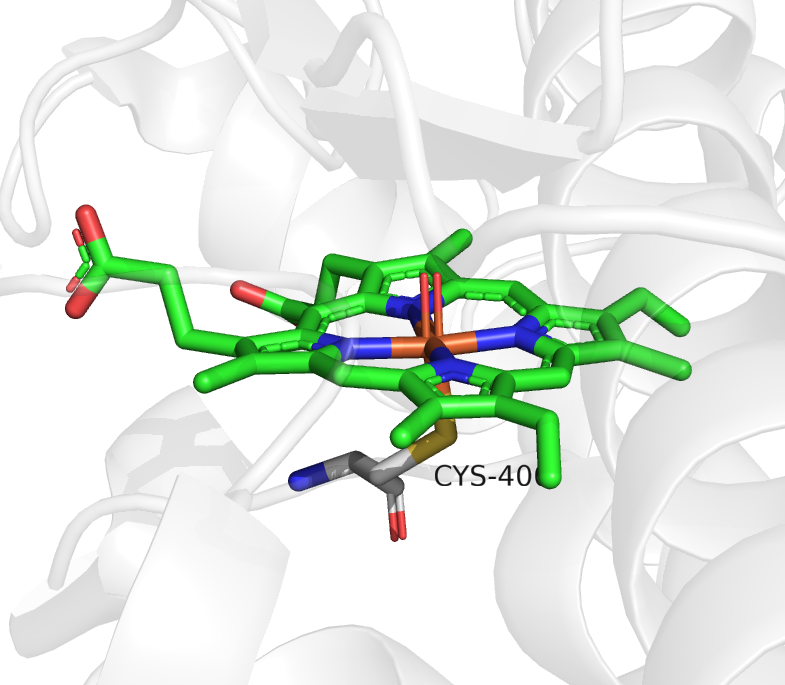
现在我们想在Fe的正上方加一个O原子,形成Fe=O,键长约1.64埃。点击pymol的菜单窗口, 右上方的Builder按钮,在chemical中选择-OCH3片段,然后点击Fe原子。关闭builder窗口。 接着删除CH3原子,点击Wizard 测量Fe和氧原子的距离,以及测量 N-Fe-O 的角度, 接着切换mouse mode为editing模式,按住ctrl键和鼠标左键移动氧原子到合适的位置就可以了。 最后再在builder中把FeO键修改为双键。首先从builder中选择双键,再点击FeO键就可以了。
调整后的效果图如下:
Q25. 请问如何ray,使颜色不变?¶
pymol 经常使用,难免会遇到一些异常情况,比如ray 颜色发生变化。
ray 之前的图片如下:

ray 之后的图片如下:
虽然清晰度变高,但是颜色失真明显。
遇到问题不要慌,先查看ray的可选参数:
help ray
PyMOL>help ray
DESCRIPTION
"ray" creates a ray-traced image of the current frame. This
can take some time (up to several minutes, depending on image
complexity).
USAGE
ray [width [,height [,antialias [,angle [,shift [,renderer [,quiet
[,async ]]]]]]]]]
ARGUMENTS
width = integer {default: 0 (current)}
height = integer {default: 0 (current)}
antialias = integer {default: -1 (use antialias setting)}
angle = float: y-axis rotation for stereo image generation
{default: 0.0}
shift = float: x-axis translation for stereo image generation
{default: 0.0}
renderer = -1, 0, 1, or 2: respectively, default, built-in,
pov-ray, or dry-run {default: 0}
async = 0 or 1: should rendering be done in a background thread?
EXAMPLES
ray
ray 1024,768
ray renderer=2
NOTES
Default width and height are taken from the current viewpoint. If
one is specified but not the other, then the missing value is
scaled so as to preserve the current aspect ratio.
angle and shift can be used to generate matched stereo pairs
renderer = 1 uses PovRay. This is Unix-only and you must have
"povray" in your path. It utilizes two two temporary files:
"tmp_pymol.pov" and "tmp_pymol.png".
See "help faster" for optimization tips with the builtin renderer.
See "help povray" for how to use PovRay instead of PyMOL's
built-in ray-tracing engine.
PYMOL API
cmd.ray(int width, int height, int antialias, float angle,
float shift, int renderer, int quiet, int async)
SEE ALSO
draw, png, save
我们可以看到examples中有一个renderer参数,试试看。
如果感觉还不行的话,再试试快速ray的命令dray。
Draw creates an oversized and antialiased OpenGL image using the current window.
It's like Ray but not ray traced. Also,
as now with Ray the oversized images are scaled and shown in the viewer window.
As Draw doesn't ray trace the shadows of the scene, it is far faster than ray.
draw 后的效果图:
Q26. select+save 和 creat+save的区别¶
一个pdb文件中有2个小分子,希望保存成sdf数据库,该数据库中有2个小分子,则使用select+save组合。
示例代码:
from pymol import cmd
cmd.load("target.pdb")
cmd.load("receptor.pdb")
cmd.select("ligand","organic")
cmd.save("ligs.sdf","ligand")
cmd.quit()
一个pdb文件中有2个小分子,希望保存成sdf数据库,该数据库中有1个小分子包含2个片段,则使用create+save组合。
示例代码:
from pymol import cmd
cmd.delete("all")
cmd.load("target.pdb")
cmd.load("receptor.pdb")
cmd.create("ligand","organic")
cmd.save("ligsssss.sdf","ligand")
cmd.quit()
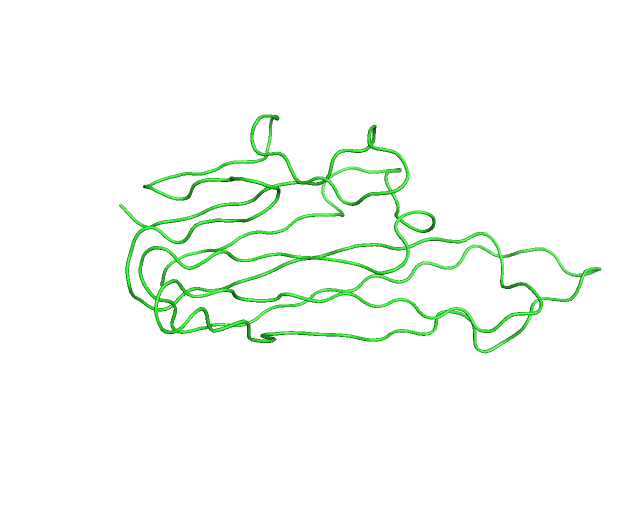
Q28. 如何读取gro格式的同源二聚体蛋白¶
分子动力学中最重要的索引是原子编号, 在处理过程中会忽略chain的编号。 比如原来是A(res id: 1-100)和B(res id:1-200,原子编号)的两条链, 在处理过程中会自动去掉chainID.
PYMOL 打开只显示一条chain。如下所示:
然后切换到molecular 模式下,选中分子,分配chain ID 就可以。
PyMOL>alter sele,chain='A'
Alter: modified 2322 atoms.
PyMOL>sort
You clicked /TMD_close_open//X/ASP`1/CA
Selector: selection "sele" defined with 2322 atoms.
PyMOL>alter sele,chain='B'
Alter: modified 2322 atoms.
PyMOL>sort
修改后:
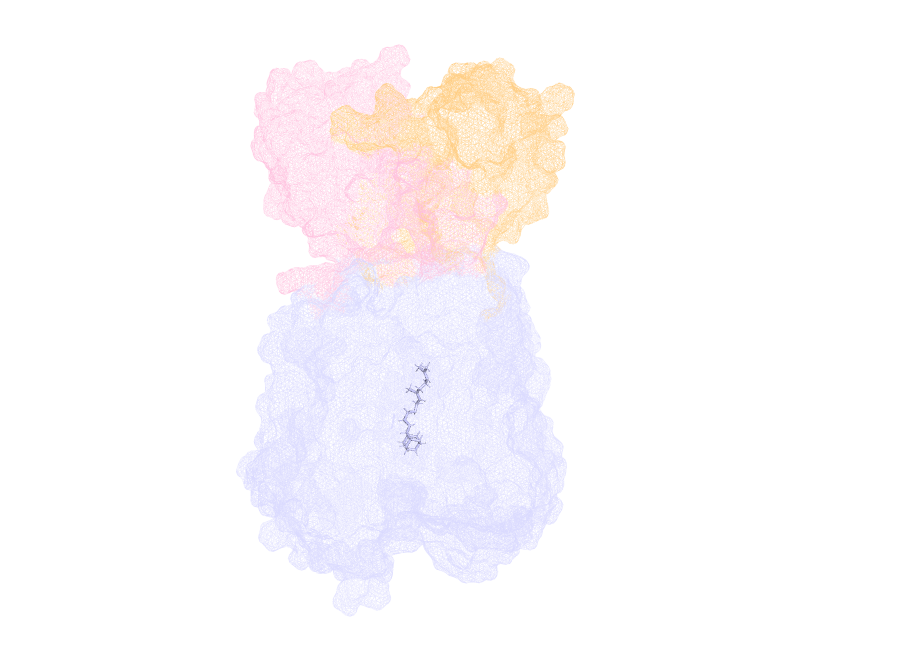
Q29. H原子作为独立的sequence问题¶
以8FD7的前3个氨基酸为例, 对蛋白进行加H后, H原子会单独称为一个Sequence如下所示:
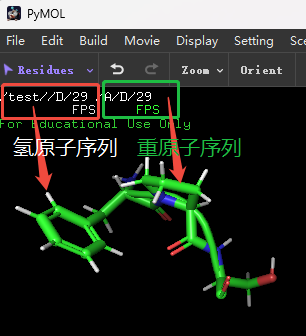
为什么会出现这样的原因:回到PDB网站上我们可以看到8FD7的chainA有2个编号:
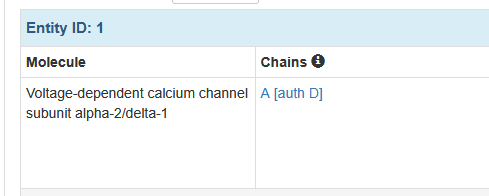
若PDB将作者的链D重命名为A,则显示为:A [auth D]
auth 是author的缩写。
回到PDB文本文件如下:
ATOM 1 N PHE D 29 95.971 29.422 51.903 1.00 95.34 A N
ATOM 2 CA PHE D 29 95.755 30.290 53.053 1.00 95.24 A C
ATOM 3 C PHE D 29 96.463 29.730 54.284 1.00 98.76 A C
ATOM 4 O PHE D 29 96.309 28.554 54.610 1.00100.02 A O
ATOM 5 CB PHE D 29 94.259 30.455 53.327 1.00 96.13 A C
ATOM 6 CG PHE D 29 93.909 31.725 54.048 1.00100.34 A C
ATOM 7 CD1 PHE D 29 93.986 31.802 55.429 1.00 98.76 A C
ATOM 8 CD2 PHE D 29 93.498 32.844 53.342 1.00100.69 A C
ATOM 9 CE1 PHE D 29 93.664 32.971 56.091 1.00 99.15 A C
ATOM 10 CE2 PHE D 29 93.175 34.015 53.997 1.00100.75 A C
ATOM 11 CZ PHE D 29 93.258 34.079 55.373 1.00100.35 A C
ATOM 12 N PRO D 30 97.245 30.568 54.959 1.00 94.90 A N
ATOM 13 CA PRO D 30 97.981 30.098 56.137 1.00 94.06 A C
ATOM 14 C PRO D 30 97.049 29.754 57.289 1.00 90.82 A C
ATOM 15 O PRO D 30 95.944 30.287 57.413 1.00 91.57 A O
ATOM 16 CB PRO D 30 98.886 31.285 56.487 1.00 94.01 A C
ATOM 17 CG PRO D 30 98.192 32.466 55.919 1.00 91.52 A C
ATOM 18 CD PRO D 30 97.506 31.988 54.672 1.00 92.23 A C
ATOM 19 N SER D 31 97.515 28.843 58.139 1.00 87.42 A N
ATOM 20 CA SER D 31 96.723 28.390 59.271 1.00 90.71 A C
ATOM 21 C SER D 31 96.633 29.482 60.337 1.00 91.76 A C
ATOM 22 O SER D 31 97.320 30.507 60.283 1.00 92.05 A O
ATOM 23 CB SER D 31 97.317 27.113 59.862 1.00 90.84 A C
ATOM 24 OG SER D 31 98.653 27.320 60.282 1.00 91.19 A O
我们可以看到PDB系统在73列增加一个chain的别名A 。
直接准备后的文件,如下:
ATOM 1 N PHE D 29 95.797 29.420 51.882 1.00 95.34 A N
ATOM 2 CA PHE D 29 95.585 30.383 52.980 1.00 95.24 A C
ATOM 3 C PHE D 29 96.331 29.834 54.234 1.00 98.76 A C
ATOM 4 O PHE D 29 96.127 28.670 54.592 1.00100.02 A O
ATOM 5 CB PHE D 29 94.075 30.603 53.256 1.00 96.13 A C
ATOM 6 CG PHE D 29 93.723 31.827 54.089 1.00100.34 A C
ATOM 7 CD1 PHE D 29 93.804 31.781 55.497 1.00 98.76 A C
ATOM 8 CD2 PHE D 29 93.454 33.062 53.461 1.00100.69 A C
ATOM 9 CE1 PHE D 29 93.576 32.927 56.245 1.00 99.15 A C
ATOM 10 CE2 PHE D 29 93.210 34.194 54.229 1.00100.75 A C
ATOM 11 CZ PHE D 29 93.270 34.127 55.615 1.00100.35 A C
ATOM 12 H1 PHE D 29 95.181 29.605 51.104 1.00 95.34 H
ATOM 13 H2 PHE D 29 96.743 29.459 51.528 1.00 95.34 H
ATOM 14 HA PHE D 29 95.993 31.333 52.639 1.00 95.24 H
ATOM 15 HB3 PHE D 29 93.639 29.720 53.728 1.00 96.13 H
ATOM 16 HB2 PHE D 29 93.547 30.702 52.306 1.00 96.13 H
ATOM 17 HD1 PHE D 29 94.047 30.855 55.997 1.00 98.76 H
ATOM 18 HD2 PHE D 29 93.422 33.130 52.383 1.00100.69 H
ATOM 19 HE1 PHE D 29 93.643 32.884 57.322 1.00 99.15 H
ATOM 20 HE2 PHE D 29 92.980 35.133 53.747 1.00100.75 H
ATOM 21 HZ PHE D 29 93.088 35.014 56.204 1.00100.35 H
ATOM 22 N PRO D 30 97.184 30.661 54.888 1.00 94.90 A N
ATOM 23 CA PRO D 30 97.975 30.223 56.057 1.00 94.06 A C
ATOM 24 C PRO D 30 97.121 29.980 57.318 1.00 90.82 A C
ATOM 25 O PRO D 30 96.208 30.757 57.598 1.00 91.57 A O
ATOM 26 CB PRO D 30 98.985 31.364 56.250 1.00 94.01 A C
ATOM 27 CG PRO D 30 98.279 32.596 55.721 1.00 91.52 A C
ATOM 28 CD PRO D 30 97.471 32.058 54.548 1.00 92.23 A C
ATOM 29 HA PRO D 30 98.514 29.305 55.813 1.00 94.06 H
ATOM 30 HB3 PRO D 30 99.875 31.167 55.651 1.00 94.01 H
ATOM 31 HB2 PRO D 30 99.310 31.495 57.280 1.00 94.01 H
ATOM 32 HG3 PRO D 30 98.959 33.403 55.449 1.00 91.52 H
ATOM 33 HG2 PRO D 30 97.595 32.971 56.482 1.00 91.52 H
ATOM 34 HD2 PRO D 30 96.572 32.657 54.396 1.00 92.23 H
ATOM 35 HD3 PRO D 30 98.065 32.086 53.633 1.00 92.23 H
ATOM 36 N SER D 31 97.441 28.891 58.041 1.00 87.42 A N
ATOM 37 CA SER D 31 96.748 28.429 59.251 1.00 90.71 A C
ATOM 38 C SER D 31 96.732 29.454 60.403 1.00 91.76 A C
ATOM 39 O SER D 31 97.644 30.276 60.509 1.00 92.05 A O
ATOM 40 CB SER D 31 97.353 27.075 59.690 1.00 90.84 A C
ATOM 41 OG SER D 31 98.617 27.209 60.312 1.00 91.19 A O
ATOM 42 H SER D 31 98.207 28.312 57.729 1.00 87.42 H
ATOM 43 HA SER D 31 95.713 28.249 58.953 1.00 90.71 H
ATOM 44 HB3 SER D 31 97.439 26.395 58.841 1.00 90.84 H
ATOM 45 HB2 SER D 31 96.686 26.590 60.403 1.00 90.84 H
ATOM 46 HG SER D 31 99.270 27.425 59.641 1.00 91.19 H
我们可以看到,软件并不会为H原子增加chain的别名A。
处理方式: 清空col73列,或者col73增加别名A。 个人倾向于清空的处理方式。
修改后的效果如下: